
SCRIPPS RESEARCH INSTITUTE
Scripps Research analysis of public genome sequence data for SARS-CoV-2 and related viruses found no evidence that the virus was produced in a laboratory or was engineered
The new SARS-CoV-2 coronavirus that emerged in the city of Wuhan, China, last year and has since caused a large-scale COVID-19 epidemic and spread to more than 70 countries, is a product of natural evolution , according to results published in the journal Nature Medicine .
Analysis of public genome sequence data for SARS-CoV-2 and related viruses found no evidence that the virus was produced in a laboratory or otherwise engineered.
"By comparing the available genome sequence data for known coronavirus strains, we can firmly determine that SARS-CoV-2 originated through natural processes," said Kristian Andersen, PhD, associate professor of immunology and microbiology at Scripps Research and corresponding author on the paper.
In addition to Andersen, authors of the paper, "The proximate origin of SARS-CoV-2," include Robert F. Garry of Tulane University; Edward Holmes, University of Sydney; Andrew Rambaut, from the University of Edinburgh; W. Ian Lipkin, Columbia University.
Coronaviruses are a large family of viruses that can cause diseases that vary widely in severity. The first known serious illness caused by a coronavirus emerged with the 2003 Severe Acute Respiratory Syndrome (SARS) epidemic in China. A second outbreak of serious illness began in 2012 in Saudi Arabia with Middle East Respiratory Syndrome (MERS).
On December 31 last year, Chinese authorities alerted the World Health Organization about an outbreak of a new strain of coronavirus that causes severe illness, which was later named SARS-CoV-2. As of February 20, 2020, nearly 167,500 cases of COVID-19 have been documented, although many milder cases have likely gone undiagnosed. The virus has killed more than 6,600 people.
Shortly after the epidemic began, Chinese scientists sequenced the SARS-CoV-2 genome and made the data available to researchers around the world.
The resulting genomic sequence data has shown that Chinese authorities quickly detected the epidemic and that the number of COVID-19 cases has increased due to human-to-human transmission after a single introduction into the human population. Andersen and her collaborators at several other research institutions used this sequencing data to explore the origins and evolution of SARS-CoV-2 by focusing on several telltale features of the virus.
The scientists analyzed the genetic template for spike proteins , armor on the outside of the virus that it uses to trap and penetrate the outer walls of human and animal cells. More specifically, they focused on two important features of the spike protein: the receptor-binding domain (RBD), a type of grappling hook that attaches to host cells, and the cleavage site, a molecular can opener that allows the virus to break open and enter host cells.
Evidence of natural evolution
The scientists discovered that the RBD portion of the SARS-CoV-2 spike proteins had evolved to effectively target a molecular feature on the outside of human cells called ACE2 , a receptor involved in regulating blood pressure. The SARS-CoV-2 spike protein was so effective at binding to human cells, in fact, that scientists concluded it was the result of natural selection and not the product of genetic engineering .
This evidence of natural evolution was supported by data on the backbone of SARS-CoV-2: its overall molecular structure. If someone were looking to engineer a new coronavirus as a pathogen, they would have built it from the backbone of a virus that is known to cause disease. But scientists found that the backbone of SARS-CoV-2 differed substantially from those of already known coronaviruses and mostly resembled related viruses found in bats and pangolins.
"These two features of the virus, mutations in the RBD portion of the spike protein and its distinct backbone, rule out laboratory manipulation as a possible origin of SARS-CoV-2," Andersen said.
Josie Golding, PhD, epidemic leader at the U.K.-based Wellcome Trust, said Andersen and her colleagues’ findings are "crucially important in providing an evidence-based view of the rumors that have been circulating about the origins." of the virus (SARS-CoV)-2) causing COVID-19."
"They conclude that the virus is a product of natural evolution," adds Goulding, "ending any speculation about deliberate genetic engineering."
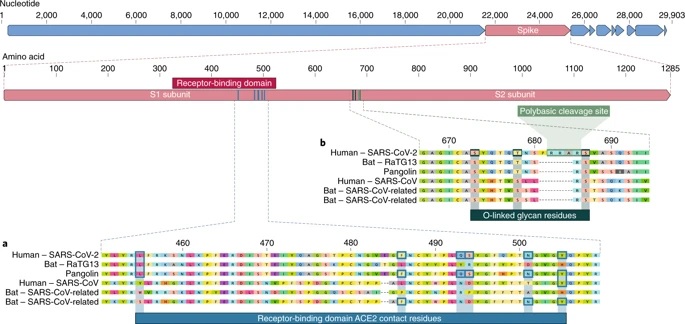
a, Mutations in the contact residues of the SARS-CoV-2 spike protein. The SARS-CoV-2 spike protein (red bar at top) was aligned against the more closely related SARS-CoV-like coronaviruses and SARS-CoV itself. Key residues on the spike protein that contact the ACE2 receptor are marked with blue boxes in both SARS-CoV-2 and related viruses, including SARS-CoV (Urbani strain). b, Acquisition of the polybasic cleavage site and O-linked glycans. Both the polybasic cleavage site and the three adjacent predicted O-linked glycans are unique to SARS-CoV-2 and were not previously seen in B-lineage betacoronaviruses. Sequences shown are from NCBI GenBank, accession codes MN908947, MN996532, AY278741, KY417146, and MK211376. The pangolin coronavirus sequences are a consensus generated from SRR10168377 and SRR10168378 (NCBI BioProject PRJNA573298) 29,30.
Possible origins of the virus
Based on their genomic sequencing analysis, Andersen and his collaborators concluded that the most likely origins for SARS-CoV-2 followed one of two possible scenarios.
In one scenario, the virus evolved to its current pathogenic state through natural selection in a non-human host and then jumped to humans . This is how previous coronavirus outbreaks have emerged, with humans contracting the virus after direct exposure to civets (SARS) and camels (MERS). The researchers proposed bats as the most likely reservoir for SARS-CoV-2, as it is very similar to a bat coronavirus. However, there are no documented cases of direct bat-human transmission, suggesting that an intermediate host was probably involved between bats and humans.
In this scenario, the two distinguishing features of the SARS-CoV-2 spike protein, the RBD portion that binds to cells and the cleavage site that opens the virus, would have evolved to their current state before entering humans. . In this case, the current epidemic would likely have arisen quickly as soon as humans were infected, as the virus would have already developed the characteristics that make it pathogenic and capable of spreading between people.
In the other proposed scenario, a non-pathogenic version of the virus jumped from an animal host to humans and then evolved to its current pathogenic state within the human population. For example, some coronaviruses from pangolins, armadillo-like mammals found in Asia and Africa, have an RBD structure very similar to that of SARS-CoV-2. A coronavirus from a pangolin could have been transmitted to a human, either directly or through an intermediate host, such as civets or ferrets.
So the other characteristic spike protein of SARS-CoV-2, the cleavage site, could have evolved within a human host, possibly through limited undetected circulation in the human population before the start of the epidemic.
The researchers found that the SARS-CoV-2 cleavage site appears similar to the cleavage sites of bird flu strains that have been shown to spread easily between people. SARS-CoV-2 could have developed such a virulent cleavage site in human cells and soon started the current epidemic, as the coronavirus would have possibly become much more capable of spreading between people.
Study co-author Andrew Rambaut cautioned that it is difficult, if not impossible, to know at this point which scenario is most likely. If SARS-CoV-2 entered humans in its current pathogenic form from an animal source, the likelihood of future outbreaks increases, as the strain of virus causing the disease could still be circulating in the animal population and could re-emerge. jump humans . The chances are lower that a non-pathogenic coronavirus will enter the human population and then develop properties similar to SARS-CoV-2.















