
| 1. Introduction |
Melanocytes originate from neural crest progenitors during embryonic development and are pigmented cells that produce melanin as a photoprotective response. Although most commonly found in the skin and hair, melanocytes are also found on mucosal surfaces and in the uveal tract of the eye. Clonal proliferation of melanocytes in these respective anatomical regions gives rise to cutaneous, mucosal, and uveal melanomas.
The most important risk factors for cutaneous melanomas include chronic exposure to ultraviolet (UV) rays in older adults and blistering sunburns during adolescence and childhood; this is supported by the UV-related C > T nucleotide substitutions that predominate in the genetics of cutaneous melanomas. At the molecular level, this malignant transition from melanocytes to melanoma is characterized and apparently depends on the constitutive activation of the RAS-RAF-MAPK/ERK pathway, which regulates cell proliferation, invasion, angiogenesis and metastasis.
The identification of driver mutations, such as BRAFv600E/K, along this pathway has led to the development of targeted therapy with dual inhibition of BRAF and MEK, which has contributed to a notable improvement in the overall survival of melanoma patients. advanced BRAF V600 ( Figure 1 ).
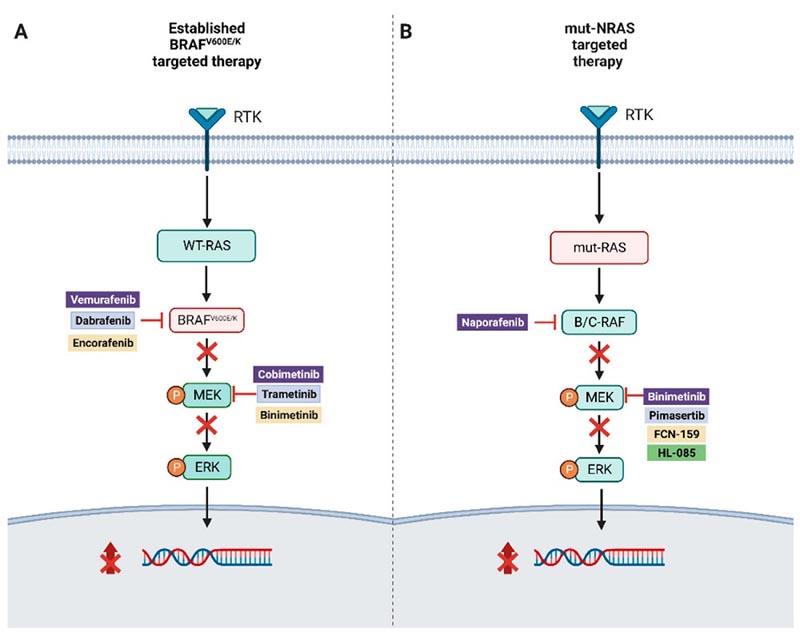
Figure 1.
( A ) Illustration of combined targeted therapy in BRAFv600E/K-mutated melanoma.
( B ) Targeted therapy approaches for NRAS-mutated melanoma.
The therapeutic landscape for melanoma has come a long way from the days when the prognosis for patients with advanced metastatic melanoma was measured in months with dacarbazine and interleukin-2 as the only FDA-approved treatment options. The emergence of immune checkpoint-based immunotherapy as a first-line treatment has significantly changed outcomes. In this review, the authors highlight recent advances in targeted therapies for melanoma.
| 2. RAF mutations, via MAPK |
In melanoma, the molecular targets lie largely within the MAPK signaling pathway, which appears critical for melanoma progression. The pathway regulates a wide range of cellular activities, including cell proliferation, survival, angiogenesis, and migration. The RAF protein family consists of three enzymes: ARAF, BRAF, and CRAF (RAF-1). Mutations are present in approximately 8% of human tumors, with BRAF-activating somatic mutations in 40-66% of melanomas.
| 3. BRAF mutant resistance |
Acquired resistance with clinical relapse is almost uniform in all patients treated with the BRAF/MEK inhibitor combination. This is partly due to numerous drug resistance mechanisms, which make it difficult to prevent or treat effectively.
| 4. Pan-RAF Inhibitors |
Pan-RAF inhibitors have been developed as an alternative approach to prevent or overcome resistance and paradoxical activation of specific BRAF inhibitors in BRAF-WT- and NRAS-mutated patients, as well as in the subset of BRAF V600E mutated patients with resistance. acquired.
Since early studies showed limited clinical efficacy of pan-RAF monotherapy, resistance mechanisms were elucidated using melanoma cells that were treated and eventually developed resistance to belvarafenib, a pan-RAF inhibitor. In vitro, belvarafenib-resistant clones were found to have reactivation of the MAPK pathway through mutations in ARAF leading to dimerization and kinase activity. In vivo, ctDNA obtained from patients with progressive disease after belvarafenib treatment was also found to have ARAF mutations.
| 5. NRAS |
NRAS is one of three proteins in the RAS protein family, which are in their active form when bound to GTP and in their inactive form when bound to GDP. Activated RAS protein signals through the MAP kinase cascade to promote proliferative and antiapoptotic activities. NRAS is the second most common driver mutation in cutaneous melanoma, second only to BRAF, and is found in 25 to 30% of cases. These mutations frequently occur in exon 2, leading to an AA substitution at position 61 (glutamine to arginine, lysine, or histidine).32]. The most common oncogenic mutations in RAS interfere with the return of RAS to an inactive GDP-bound state, which is catalyzed by GTPase-activating proteins.
NRAS-mutated patients with advanced melanoma at diagnosis have significantly shorter overall survival (15.5 months vs. 23.5 months). Despite its prevalence and importance in melanoma, RAS has historically not been shown to be amenable to direct treatment due to the lack of an easily pharmacological treatment. GTP-competitive drugs are limited by the extremely high affinity of RAS for GTP and high intracellular concentrations of GTP.
| 6. ERK |
ERK, a downstream effector of the MAPK pathway, catalyzes the phosphorylation of nuclear transcription factors once it is in its active state. ERK signaling in melanoma becomes independent of feedback mechanisms due to mutations in BRAF and NRAS. Furthermore, ERK signaling is maintained without hyperactivation and subsequent cell cycle arrest or death due to intrinsic feedback regulation through DUSP transcription. ERK signaling is restored in patients who acquire resistance to RAF and MEK inhibition, generating interest in directly targeting ERK to address resistance.
| 7. Atypical BRAF mutations |
Melanomas with atypical BRAF mutation encompass a diverse genetic alteration and are often subdivided into V600 and non-V600 mutants. Each mutation is classified into classes I, II or III based on its signaling mechanism and kinase activity, leading to different degrees of molecular deregulation. The rare mutations, BRAF V600 including V600R/D/M/L, are kinase-activating monomers characteristically found in older male patients with a history of chronic sun damage. Non-V600 mutants can be classified as class II (e.g., L597P/Q/R/S, K601E, G469R/S/A) or III (G596R, D594Y/N/G/E, D287Y) depending on the formation of dimers to activate RAF kinases or heterodimers that alter kinase activity completely, resulting in paradoxical activation of ERK signaling, respectively.
Typically, class II and III tumors have a more aggressive clinical course and are associated with a worse prognosis. Atypical BRAF mutations are most commonly seen in mucosal melanoma.
| 8. Inhibition of MEK CDK4/6 + |
Fifty percent of NRAS mutant melanomas have genetic aberrations in cell cycle-associated genes, providing a rationale for combining MEK inhibitors with CDK4/6 inhibitors. CDK4/6 inhibitors cause cell cycle arrest by preventing the CDK4/6-cyclin D1 complex from phosphorylating the retinoblastoma protein. A preclinical study using NRAS mutant human melanoma cell lines revealed synergistic effects on both apoptosis and cell cycle arrest with corresponding tumor regression. A Phase II trial evaluating the efficacy of the pan-RAF inhibitor naporafenib combined with ribociclib in patients with metastatic or unresectable NRAS-mutated melanoma is currently underway.
| 9 . SHP2 |
Protein tyrosine phosphatase 2, or SHP2, has recently been the basis for new targeted therapy approaches. SHP2 functions as a protein tyrosine phosphatase involved in the activation of several signaling pathways, including RAS/Raf/MAPK, PI3K/ AKT, JAK/STAT, and PD-1/PD-L1. Consequently, modulation of SHP2 activity may affect cell survival and immune regulatory pathways.
| 10. Autophagy |
Melanomas that have acquired resistance to BRAF plus MEK inhibition are often characterized by rapidly progressive disease. One of the resistance mechanisms that has been identified is autophagy, an adaptive cellular process in which abnormal proteins, damaged organelles, and pathogens are broken down through lysosomal degradation to recycle cellular components into molecular precursors and energy to facilitate self-renewal.
Specifically, MEK1/2 inhibition increases autophagic flux by activating the LKB1-AMPK-ULK1 signaling axis in RAS-driven cancer cells. Therefore, autophagy has become a rational target to overcome BRAF/MEK resistance.
| 11 . Epigenetic targets |
EZH2 encodes a catalytic subunit of Polycomb repressive complex 2 (PRC2), which facilitates trimethylation of histone H3 lysine 27. PRC2 plays a key role in epigenic regulation by promoting chromatin compaction. EZH2 is highly expressed in human primary and metastatic melanoma and is considered necessary for melanogenesis, proliferation, and metastasis; its expression is associated with a worse prognosis. Point mutations of EZH2 result in a gain of function in 5% of melanomas and are often found in coexistence with BRAF activators that may indicate an oncogenic synergism.
| 12. BAP1 and HDAC |
BRCA1-associated protein 1 (BAP1) is a tumor suppressor gene that is frequently mutated in metastatic uveal melanomas in the late stages of melanomagenesis. The mutation produces a loss of gene function and drives the cells to dedifferentiate and acquire stem-like properties with greater capacity for self-replication. Alterations in epigenetic regulations contribute to this phenotype, as demonstrated by the restoration of melanocyte differentiation when uveal melanoma cells with BAP1 mutations are treated with histone deacetylase (HDAC) inhibitors. However, the clinical efficacy of HDAC inhibitors has not been established.
| 13. Enhanced immunotherapy |
The advent of immunotherapy has represented the most significant advance in the care of patients with advanced melanoma since initial efforts with systemic therapy many years ago. Although median survival has been reported to be as high as 72.1 months in patients treated with the ipilimumab/nivolumab combination, this approach may be limited by significant treatment-related immunological adverse events and the development of resistance.
Several novel approaches have been undertaken to combine immunotherapy with targeted therapy agents to overcome resistance and improve outcomes. A well-established approach has been to reverse the immunosuppressive effect of proangiogenic factors in the tumor microenvironment using tyrosine kinase inhibitors that interfere with VEGF signaling to increase the antitumor efficacy of PD-1/L1 inhibitors.
| 14.Conclusions |
The therapeutic landscape for advanced melanoma has experienced significant expansion in recent decades. Despite advances in immunotherapy and targeted therapies that have revolutionized the melanoma treatment paradigm, there is a persistent need to develop novel therapies, particularly for patients who have progressed or for those patients who are not good candidates for system-based therapies. immunological.
The identification of driver mutations for the rational design of new targeted therapies will be of vital importance to continue optimizing clinical outcomes and achieve the difficult cure of melanoma.















