| Pharmacodermies Par Ana Clara Torre (Argentine). |
Tout effet nocif ou indésirable apparaissant après l’utilisation d’un médicament à dose prophylactique, diagnostique ou thérapeutique. L’OMS estime qu’elle représente environ 15 % de tous les effets indésirables des médicaments.
Ils peuvent être classés en simples ou non compliqués (90 à 95 % des cas), dans lesquels on observe une atteinte cutanée légère, sans atteinte systémique, et complexes ou compliqués (2 à 5 % des cas), qui présentent une atteinte. atteinte cutanée et/ou systémique sévère. Dans ce dernier groupe, on observe généralement des altérations biologiques, un signe de Nikolsky, des pustules généralisées, des lésions cibles, des lésions purpuriques ou une érythrodermie.
En fonction de la gravité de la maladie, ils peuvent être traités de la manière suivante :
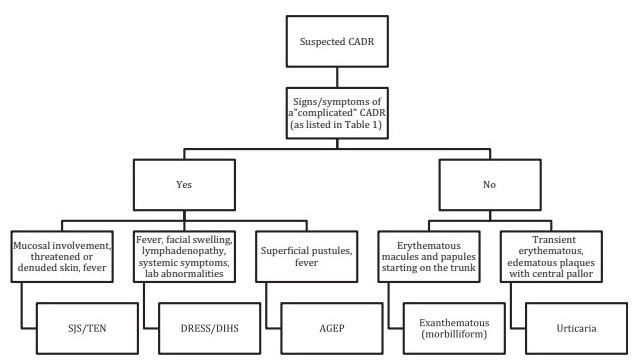
CADR : effet indésirable cutané au médicament
AGEP : éruption pustuleuse aiguë généralisée
SJS/TEN : syndrome de Steven-Johnson/nécrolyse épidermique toxique
DRESS/DIHS : effet indésirable au médicament avec éosinophilie et symptômes systémiques/syndrome d’hypersensibilité médicamenteuse.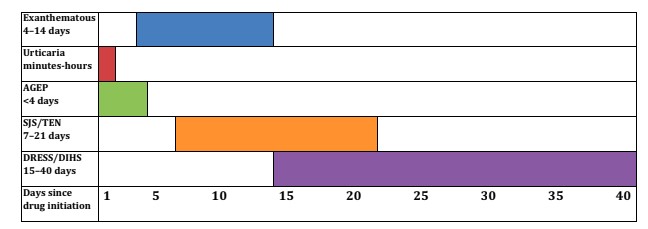
Il est à noter que tous les symptômes ne présentent pas le même temps de latence après l’introduction du médicament, ce qui oblige à s’interroger même sur ceux consommés jusqu’à 40 jours avant l’apparition de l’affection cutanée.
Swanson L, Colven RM. Approche du patient présentant une réaction cutanée indésirable présumée au médicament. Med Clin Nord Am. nov. 2015;99(6):1337-48
Concernant l’étude de ces conditions cliniques, une étude en laboratoire est essentielle, à la fois pour évaluer les lésions des organes cibles et pour exclure d’autres diagnostics différentiels.
| Mastocytose infantile Par le Prof. Dr. Antonio Torrelo (Espagne). |
Ensemble de maladies malignes dans lesquelles une accumulation pathologique de mastocytes est observée dans différents tissus. Elles sont dues à une mutation clonale du gène kit. Elle peut débuter à des âges différents et présenter des phénotypes cliniques différents, ce qui rend sa classification plus complexe.
L’OMS ne reconnaît que 3 sous-types chez les patients pédiatriques en fonction du nombre de lésions, laissant de côté certains paramètres tels que la génétique, la morphologie, l’histologie ou le comportement de la maladie. Cette classification génère beaucoup de controverses, utilisant des termes ambigus, ne permettant souvent pas de classer les patients que nous évaluons quotidiennement.
Torrelo, compte tenu de son expérience sur ce sujet, propose 2 types de mastocytose infantile , les types 1 et 2. Le premier, ou classique, est le type observé chez l’adulte. Type 2 ou bien différencié, fréquemment observé chez l’enfant.
Type 1 : petites lésions cutanées mélaniques, maculaires ou papuleuses ; nombre variable de blessés. La dermoscopie montre des télangiectasies. Signe Darier + mais difficile à provoquer. Des ampoules peuvent parfois être observées. L’histopathologie montre des mastocytes périvasculaires en nombre modéré. Elle s’accompagne rarement de symptômes systémiques. Mastocytes avec mutation du gène kit au niveau du codon 815 ou 816. Les mastocytes de la moelle osseuse sont en forme de fuseau et marquent positivement CD2 et CD25 en cytométrie en flux. Il ne répond pas à l’imatinib mais répond à la midostaurine.
C’est la variété la plus observée dans l’enfance.
Type 2 : macules, plaques, papules, gros nodules et il peut y avoir un grand nombre de lésions. Des ampoules peuvent souvent être observées. Avec l’évolution, des formes anétodermiques et érythrodermiques peuvent être observées. Signe Darier + bien visible. L’histopathologie montre des mastocytes ronds et granulés, avec des noyaux polylobulés. Les symptômes systémiques sont fréquents. Les mastocytes de la moelle osseuse marquent négativement en cytométrie en flux pour CD2 et CD25. Les mutations du kit ne sont pas observées dans le codon 815 ou 816, il répond très bien à l’inhibiteur de la tyrosine kinase imatinib. Cette variété correspond à 5 à 10 % des cas de mastocytose observés à l’âge adulte, puisqu’elle a tendance à disparaître.
| Dermatite atopique discoïde et lichénoïde Par le Prof. Dr. José Ollague (Équateur). |
Ollague propose (sur la base d’une série de patients évalués dans son cabinet) d’envisager une sous-variété de dermatite atopique (DA) chez les patients présentant des plaques discoïdes et lichénoïdes , situées symétriquement dans les zones d’extension des membres, respectant les flexions , accompagnées d’hyperkératose. palmo-plantaire. De plus, les Ig E sont très riches en sérum, ce qui se traduit cliniquement par des démangeaisons intenses . L’histopathologie de ces patients met en évidence la présence de modifications psoriasiformes dans l’épiderme et d’éosinophiles dans le derme .
Pour différencier la MA discoïde et lichénoïde du syndrome de Sulzberger-Garbe, cette sous-variété est proposée comme une entité indépendante.
| Inhibiteurs de JAK dans la dermatite atopique Par le Dr Paula Luna (Argentine). |
La MA présente une physiopathogenèse très complexe, résultat d’une interaction de multiples acteurs. Il a certainement une base génétique, un pool de gènes, liés à la barrière cutanée, au prurit et à l’immunité. En revanche, il est important de mettre en évidence le rôle du microbiome tant dans la genèse que dans la perpétuité de la MA.
On sait aujourd’hui que l’origine de la MA ne réside pas uniquement dans la voie Th2 mais aussi dans la voie Th1, avec de multiples cytokines impliquées en plus des bien connues 4 et 13 (inhibées par le dupilumab) ; IL 31 responsable du prurit, entre autres.
Dans la pratique clinique, nous observons différents phénotypes de MA, avec des différences observées en fonction de la race, du temps d’évolution et du groupe d’âge, entre autres facteurs en question. Cela pourrait correspondre aux voies inflammatoires impliquées dans chaque cas particulier. Le traitement de cette pathologie évolue vers un paradigme « sur mesure », adaptant le traitement à chaque patient en fonction du ou des parcours concernés. La grande hétérogénéité clinique et physiopathogène signifie qu’il existe de multiples options thérapeutiques, d’efficacité variable, depuis les inhibiteurs cellulaires jusqu’aux inhibiteurs hautement spécifiques tels que les inhibiteurs biologiques.
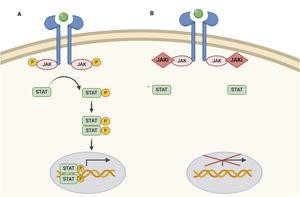
Les inhibiteurs de JAK sont modérément spécifiques, inhibant plusieurs cytokines le long de la voie JAK-STAT. Ils empêchent la phosphorylation de STAT, son activation et donc son action au niveau du noyau cellulaire en modulant la transcription génétique.
Procès-verbal dermosifiliographique du 21 janvier 2021
JAK possède 4 sous-variétés, fonctionnelles sous forme de dimères, agissant comme récepteur de différentes cytokines et activant ainsi les voies physiologiques.
Il existe 3 inhibiteurs de JAK à usage systémique : le baricitinib, l’abrocitinib et l’upadacitinib ; Le seul approuvé en Europe pour la MA (et en cours d’approbation aux USA) est le baricitinib, sélectif pour JAK 1 et 2. Les deux autres sont sélectifs pour JAK 1. Le baricitinib et l’upadacitinib sont autorisés dans notre pays pour le traitement de la PR. Des protocoles de recherche sont actuellement en cours pour approuver le baricitinib dans le traitement de la MA chez les enfants et les adultes ; de même avec l’upacitinib. Ces 3 médicaments sont considérés comme de petites molécules, tous sont ingérés par voie orale en une seule prise quotidienne, présentant un délai d’action rapide. Jusqu’à présent, il a été constaté que les inhibiteurs de JAK apportaient un grand soulagement du prurit et très rapidement chez les patients atteints de MA, principale limitation de la qualité de vie.
Parmi les effets indésirables de classe, on peut citer les lymphomes, les thromboses et les infections, qui ont été peu rapportés dans les protocoles de recherche. Le baricitinib présente l’eczéma herpeticum et le zona comme effet indésirable ; réaction acnéiforme à l’upadacitinib, couramment observée dans les protocoles de recherche.
Ces molécules semblent constituer un groupe thérapeutique prometteur pour les patients atteints de MA.
| Le point sur le traitement des lymphomes cutanés . |
| Le point sur le traitement du mycosis fongoïde Par le Prof. Dr. José Sanches (Brésil). |
La mycosis fongoïde (MF) est une tumeur qui évolue vers un large éventail de manifestations cliniques. Initialement, elle se présente généralement sous forme de plaques à peine infiltrées, évoluant au fil du temps vers des lésions tumorales notamment infiltrées et ulcérées. Sa genèse réside dans les lymphocytes T matures et atypiques, qui touchent initialement l’épiderme et, au fur et à mesure de l’évolution de la maladie, touchent également les ganglions lymphatiques, le sang périphérique et les organes distants.
Concernant le traitement du MF, il n’existe pas de lignes directrices claires. C’est une pathologie incurable malgré les traitements dont nous disposons à ce jour.
Le traitement repose sur l’étendue des manifestations cliniques, le type de lésions présentes et la présence d’une maladie systémique. Nous disposons d’alternatives topiques et systémiques basées sur ces paramètres mentionnés précédemment. Le premier groupe comprend : les corticostéroïdes topiques, la méchloréthamine, la carmustine, le bexarotène, l’imiquimod et le resiquimode. En outre, la photothérapie UVB et PUVA, la thérapie photodynamique et la radiothérapie peuvent être mentionnées comme traitement physique.
Le resiquimode est une nouveauté dans le spectre des traitements topiques . C’est un agoniste Toll-R 7/8 très utile dans les premiers stades de la MF et présentant une très faible toxicité (irritation locale). Il se présente sous forme de gel, à 0,03 ou 0,06%. Rook et al ont prouvé qu’il y avait une amélioration significative chez 75% des patients traités, les lésions traitées s’améliorent toutes et même une régression des lésions non traitées a été observée.
Parmi les options systémiques figurent les médicaments modifiant la réponse biologique tels que l’interféron, le bexarotène, l’acitrétine, l’alemtuzumab, le vorinostate/romidepsine et le brentuximab vedotin ; agents de chimiothérapie, tels que le chlorambucil, le méthotrexate, la gemcitabine, la doxorubicine liposomale, entre autres. Ces deux derniers médicaments sont les plus utilisés en monothérapie. La photophérèse extracorporelle et la transplantation de cellules souches sont également des alternatives. Comme mentionné ci-dessus, étant donné que la MF est actuellement une tumeur incurable et en fonction de l’engagement de chaque cas particulier, les taux de réponse objective à ces traitements atteignent 30 à 50 %.
Aux stades précoces, l’interféron continue d’être le premier choix thérapeutique.
Le Brentuximab vedotin , une alternative systémique, est un anticorps monoclonal comme le mogalizumab . Ils sont étudiés dans le cadre de protocoles de recherche, mais le premier aurait apparemment un taux de rémission global de 60 %.
| Le point sur le traitement du syndrome de Sézary Par le Dr Paula Enz (Argentine). |
Suspicion clinique dans le cadre d’une érythrodermie (atteinte de plus de 80 % de la surface corporelle, T4), de ganglions lymphatiques et de cellules de Sézary dans le sang périphérique.
> Concept actuel : utilisation de thérapies multimodales dirigées vers la peau associées à des thérapies systémiques en tandem tournant (avec de nouveaux agents biologiques et immunomodulateurs). Chimiothérapie cytotoxique uniquement dans un contexte de progression de la maladie ou d’échec de la réponse clinique à un autre traitement établi.
Les traitements sont ceux mentionnés précédemment.
La photothérapie est une excellente alternative pour traiter le prurit, généralement limitant chez ces patients. De même, le bain d’électrons, qui peut être associé à des traitements topiques, à l’IFN ou à la photophérèse. Plus de 30 Gy sont efficaces, mais sont associés à une toxicité cutanée importante, ou une dose plus faible peut être utilisée dans les lésions localisées. Cela laisse la possibilité de réaliser des traitements plus courts et des retraçages.
La photophérèse est une bonne première option thérapeutique. Cela se fait 2 jours consécutifs toutes les 2, 3 ou 4 semaines. Il peut être utilisé seul ou combiné à d’autres thérapies. La réponse globale est de 40 % mais les études portent sur peu de patients. Il nécessite un système immunitaire « intact », il est donc recommandé de l’utiliser avant un traitement immunosuppresseur.
Le vorinostat et la romidepsine sont des inhibiteurs de l’acétylation des histones. Il n’existe aucune étude comparative entre les deux. Les deux ont des réponses globales d’environ 35 %.
Le mogalizumab a été approuvé en 2018, c’est un anti-CCR4 . Il a une réponse globale de 37% en SS. Il est utile en association avec d’autres traitements immunomodulateurs.
Le brentuximab est utilisé chez les patients qui présentent > 10 % de lymphocytes CD30+ dans les biopsies cutanées de patients atteints de SS.
L’alemtuzumab entraîne plusieurs effets indésirables graves (immunosuppression et infections mortelles), des protocoles sont en cours d’élaboration pour le SS à faibles doses .
L’allogreffe de moelle osseuse est une alternative chez les patients en bon état général n’ayant pas répondu au traitement de première intention. Cela peut être une alternative curative.
Monothérapie ou traitements multimodaux du SS ? De nombreuses études parlent de combiner la photophérèse avec des traitements immunomodulateurs, ce qui entraîne une augmentation de l’efficacité avec une réponse globale proche de 80 %. Un traitement multimodal serait la solution appropriée.
Enz a commenté qu’un patient atteint de SS traité par photophérèse et bexarotène a eu une infection au COVID en 2020 à un mauvais moment de son atteinte cutanée. Après l’infection, sa peau a évolué très favorablement.
> Avenir : virothérapie oncolytique, avec des vecteurs viraux de la rougeole. À l’étude, mais semble avoir reçu une bonne réponse jusqu’à présent.
| Traitement des syndromes lymphoprolifératifs cutanés primaires CD30 Par le Dr Mariana Arias (Argentine). |
Il comprend le lymphome cutané anaplasique primitif à grandes cellules (PLCLCL) et la papulose lymphomatoïde (LP). Ceux-ci représentent environ 25 % des lymphomes cutanés primitifs, deuxièmes en fréquence après le MF et le SS.
LACGCP : adultes, hommes (3 : 1), survie à 10 ans de 90 %. Tumeurs érythémateuses-violacées à croissance rapide, pouvant s’ulcérer, généralement volumineuses, dans n’importe quel site anatomique. Jusqu’à 40 % des cas peuvent présenter une régression spontanée ou récidiver. Les progressions extracutanées sont fréquentes, notamment au niveau ganglionnaire, obscurcissant le pronostic. De même, les cas de tumeurs multiples des membres inférieurs méritent une attention particulière ; car ils présentent généralement une atteinte des ganglions lymphatiques.
PL : adultes, excellent pronostic avec 100 % de survie à 10 ans. Association élevée avec d’autres lymphomes, généralement avec le MF. Cela mérite une surveillance étroite. Éruption chronique et récurrente de papules et de nodules pouvant s’ulcérer, sur le tronc et les membres. L’autoinvolution est très fréquente, entraînant des épisodes de poussées et de rémissions.
Il est essentiel d’éviter un traitement excessif car ces néoplasmes ont un très bon pronostic. Le choix est basé sur l’étendue et le nombre de blessures. Peu de preuves scientifiques des cas de récidive ou d’implication systémique.
Pour LACGCP :
> Lésion unique ou lésions groupées : ablation chirurgicale associée à une radiothérapie localisée ou radiothérapie localisée uniquement avec une marge tant latéralement qu’en profondeur de 2 cm. Avec atteinte ganglionnaire associée : radiothérapie cutanée et ganglionnaire localisée ± brentuximab ou radiothérapie cutanée et ganglionnaire localisée (1ère ligne NCCN 2021).
> Lésions multifocales : brentuximab vedotin en 1ère intention (NCCN 2021) ; Comme autres options, une association de traitements systémiques +/- traitements cutanés (topiques ou physiques) est proposée.
Revue de 10 publications de lymphomes cutanés primitifs traités par brentuximab vedotin : 100 % des patients ont présenté une réponse complète dans un délai médian de 5 semaines ; rapports d’effets indésirables légers.
Pour la LP, l’observation peut se faire sachant que le facteur limitant est la présence d’un deuxième lymphome associé.
> Lésions groupées : photothérapie ou corticoïdes topiques.
> Lésions étendues : méthotrexate, photothérapie (NCCN 2021), rétinoïdes systémiques.
| La révolution biologique dans le psoriasis Par le Prof. Dr Jonathan Barker (Royaume-Uni). |
Au cours des 30 dernières années, la compréhension du psoriasis a considérablement évolué. Dans les années 80, nous considérions sa genèse comme un trouble de la différenciation épidermique, c’est pourquoi des médicaments immunosuppresseurs tels que le méthotrexate ont commencé à être utilisés. Ensuite, l’attention s’est tournée vers l’immunologie et la cyclosporine a commencé à être utilisée ici, sur la base d’une étude suisse évaluant des patients atteints de polyarthrite rhumatoïde traités avec ce médicament. Au cours des 20 dernières années, nous avons cherché à stratifier chaque cas particulier pour proposer un traitement ciblé, sachant que les lymphocytes cutanés synthétisent un grand nombre de cytokines qui constituent un réseau complexe donnant naissance à différentes conditions cliniques. Nous nous trouvons à l’ère des traitements biologiques. Ils ont sans aucun doute modifié l’évolution du psoriasis, mais comme tout traitement, ils ont leurs limites. Ils sont très chers, ils ne sont pas efficaces dans tous les types de psoriasis, leur effet diminue avec le temps, bien sûr ils ont leurs effets indésirables, etc.
Au Royaume-Uni, les traitements biologiques sont approuvés par le National Health Service avec des contrôles stricts. Les équipes traitantes mesurent les niveaux des différents médicaments biologiques un mois après le début du traitement, car il a été constaté que les patients qui n’atteignent pas une certaine plage de concentrations souhaitée de ceux-ci dans le sérum sont candidats à suspendre le traitement biologique et à alterner le traitement pour un autre. . . Ils le font comme une stratégie pour économiser des ressources, sachant que ces patients sont plus sujets aux échecs primaires dus à un agent biologique. Barker a déclaré qu’ils avaient effectué l’adalimumab à temps.
Il est impossible de ne pas évoquer la pandémie de COVID à propos de ces patients. Barker a réalisé des enregistrements sur le suivi des patients atteints de psoriasis traités avec des produits biologiques, nous aurons donc ces résultats dans le futur. En ce qui concerne le vaccin COVID, on ne sait pas quel effet protecteur il aura sur ce groupe de patients et dans quelle mesure il est sûr. Même s’il faut être prudent avec les recommandations, il le recommande à ses patients.
Barker estime qu’au cours des 10 prochaines années, différentes options topiques émergeront et que l’utilisation de biomarqueurs dans la pratique clinique se développera, afin de catégoriser les patients et de proposer un traitement ciblé « sur mesure ».
| Nouvelles méthodes de diagnostic de la lèpre Par le Dr Heitor Goncalves (Brésil). |
Il propose des méthodes de diagnostic pour caractériser plus facilement différents défis diagnostiques de la pratique clinique : lèpre indéterminée sans altération de la sensibilité, diagnostic différentiel entre réaction inverse et récidive chez les patients paucibacillaires, diagnostic différentiel entre réaction inverse et récidive chez les patients multibacillaires, formes neuronales pures, neurones silencieux. paralysie et diagnostic précoce des patients multi- et paucibacillaires dans les zones endémiques.
> Outils de diagnostic :
Clinique LE PLUS IMPORTANT (sensibilité thermique, compromission motrice) ;
• Laboratoire : Baciloscopie (particulièrement importante chez les patients multibacillaires).
• Histopathologie (diagnostic différentiel de récidive ou de schéma réactionnel chez MB).
• Test histaminique (diagnostic différentiel des formes indéterminées, utile chez les patients pédiatriques pour les distinguer des eczématides acromiantes observées dans la MA).
• Électromyogramme : jusqu’à 40 % des patients qui ne présentent pas encore de manifestations cliniques de névrite présentent au préalable une atteinte EMG ; dd avec d’autres pathologies qui provoquent des neuropathies ; L’inconvénient est que cela ne donne pas de changements pathognomoniques.
• Echographie neuronale : utile pour évaluer l’épaississement, notamment dans les cas où une décompression est nécessaire et pour réaliser des biopsies ciblées.
• Sérologie : les anticorps anti-lèpre sont mesurés . Plus la concentration d’Ac chez le patient est élevée, plus la réponse humorale est déduite et, par conséquent, un patient multibacillaire est interprété. Utile pour surveiller les patients vivant dans des zones endémiques. La sérologie A + n’implique pas que le patient est malade, mais plutôt qu’il a été en contact avec le bacille M. leprae et justifie donc un suivi attentif. La détection se fait par méthode ELISA, avec une spécificité de 98%. De peu d’intérêt pour les patients paucibacillaires.
• PCR M. leprae : détecte les bacilles vivants ou viables, utile pour les formes neurales, indéterminées, pour différencier les phénomènes réactionnels de réinfection (il peut s’agir d’un autre type de bacille) ou de récidive (même bacille) et pour les patients paucibacillaires.
- Type conventionnel : amplifie les bacilles vivants et morts.
- Type quantitatif en temps réel.
- Transcriptase inverse : + ; signifie bacille vivant (récidive et non réaction).
• Biopsie nerveuse : nerf sensoriel ; rarement sur les nerfs moteurs (risque élevé de blessure). Dirigez-le par échographie. La PCR peut être réalisée sur le nerf.
• Etude du génome du bacille : utile pour les études de population, pour déterminer la réinfection en rechute. La possibilité de développer un vaccin est à l’étude.















