| Pharmakodermie Von Ana Clara Torre (Argentinien). |
Jede schädliche oder unerwünschte Wirkung, die nach der Einnahme eines Arzneimittels in einer prophylaktischen, diagnostischen oder therapeutischen Dosis auftritt. Die WHO schätzt, dass sie für etwa 15 % aller unerwünschten Arzneimittelwirkungen verantwortlich ist.
Sie können in einfache oder unkomplizierte Fälle (90–95 % der Fälle) eingeteilt werden, bei denen eine leichte Hautbeteiligung ohne systemische Beteiligung beobachtet wird, und in komplexe oder komplizierte Fälle (2–5 % der Fälle), bei denen eine Beteiligung vorliegt. schwere Haut- und/oder systemische Beteiligung. In dieser letzten Gruppe werden normalerweise Laborveränderungen, Nikolsky-Zeichen, generalisierte Pusteln, Zielläsionen, purpurische Läsionen oder Erythrodermie beobachtet.
Abhängig von der Schwere der Erkrankung können sie folgendermaßen behandelt werden:
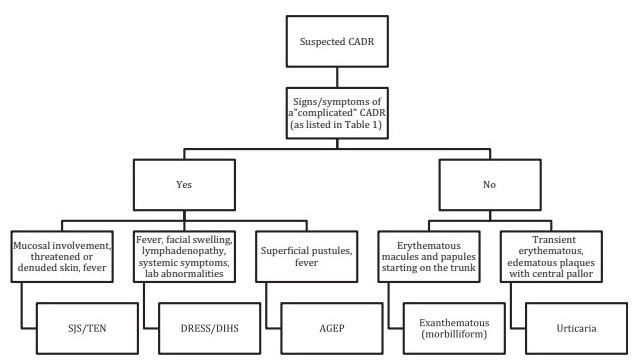
CADR: kutane unerwünschte Arzneimittelwirkung
AGEP: akuter generalisierter pustulöser Ausschlag
SJS/TEN: Steven-Johnson-Syndrom/toxische epidermale Nekrolyse
DRESS/DIHS: unerwünschte Arzneimittelwirkung mit Eosinophilie und systemischen Symptomen/medikamenteninduziertes Überempfindlichkeitssyndrom.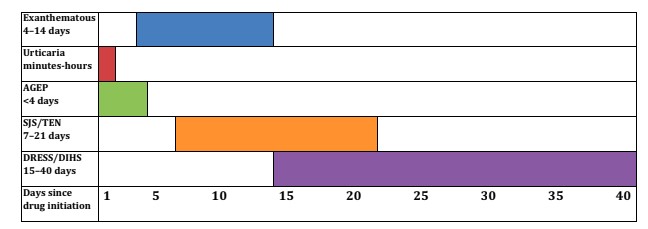
Es ist zu beachten, dass nicht alle Symptome die gleiche Latenzzeit nach der Einführung des Medikaments aufweisen, was uns dazu zwingt, selbst diejenigen in Frage zu stellen, die bis zu 40 Tage vor Auftreten der Hauterkrankung eingenommen wurden.
Swanson L, Colven RM. Umgang mit dem Patienten mit Verdacht auf eine kutane unerwünschte Arzneimittelwirkung. Med Clin North Am. 2015 Nov;99(6):1337-48
Für die Untersuchung dieser klinischen Zustände ist eine Laboruntersuchung unerlässlich, um sowohl die Schädigung des Zielorgans zu beurteilen als auch andere Differentialdiagnosen auszuschließen.
| Infantile Mastozytose Von Prof. Dr. Antonio Torrelo (Spanien). |
Reihe bösartiger Erkrankungen, bei denen eine pathologische Ansammlung von Mastzellen in verschiedenen Geweben beobachtet wird. Sie sind auf eine klonale Mutation des Kit-Gens zurückzuführen. Sie kann in unterschiedlichem Alter beginnen und unterschiedliche klinische Phänotypen aufweisen, was ihre Klassifizierung komplexer macht.
Die WHO erkennt bei pädiatrischen Patienten nur drei Subtypen anhand der Anzahl der Läsionen und lässt bestimmte Parameter wie Genetik, Morphologie, Histologie oder das Krankheitsverhalten außer Acht. Diese Klassifizierung führt zu vielen Kontroversen, da sie mehrdeutige Begriffe verwendet und es oft nicht möglich ist, die Patienten, die wir täglich untersuchen, zu klassifizieren.
Aufgrund seiner Erfahrung zu diesem Thema schlägt Torrelo zwei Arten der Mastozytose im Kindesalter vor , Typ 1 und Typ 2. Der erste oder klassische Typ ist der Typ, der bei Erwachsenen beobachtet wird. Typ 2 oder gut differenziert, häufig bei Kindern beobachtet.
Typ 1 : kleine, melanische makulöse oder papulöse Hautläsionen; unterschiedliche Anzahl von Verletzungen. Dermatoskopie zeigt Telengiektasien. Darier + Zeichen, aber schwer zu provozieren. Gelegentlich können Blasen auftreten. Die Histopathologie zeigt perivaskuläre Spindelmastzellen in mäßiger Anzahl. Es geht selten mit systemischen Symptomen einher. Mastzellen mit Mutation des Kit-Gens am Codon 815 oder 816. Die Mastzellen des Knochenmarks sind spindelförmig und markieren CD2 und CD25 in der Durchflusszytometrie positiv. Es reagiert nicht auf Imatinib, wohl aber auf Midostaurin.
Es ist die Sorte, die in der Kindheit am häufigsten beobachtet wird.
Typ 2 : Makulae, Plaques, Papeln, große Knötchen und es kann eine große Anzahl von Läsionen vorhanden sein. Häufig sind Blasen zu beobachten. Im Laufe der Evolution können Anetodermie und erythrodermische Formen beobachtet werden. Darier-Zeichen + prominent. Die Histopathologie zeigt runde und granulierte Mastzellen mit polylobulierten Kernen. Systemische Symptome sind häufig. Knochenmarksmastzellen weisen in der Durchflusszytometrie eine negative Markierung für CD2 und CD25 auf. Die Mutationen im Kit werden im Codon 815 oder 816 nicht beobachtet, es reagiert sehr gut auf den Tyrosinkinase-Inhibitor Imatinib. Diese Variante entspricht 5–10 % der im Erwachsenenalter beobachteten Fälle von Mastozytose, da sie tendenziell verschwindet.
| Atopische diskoide und lichenoide Dermatitis Von Prof. Dr. José Ollague (Ecuador). |
Ollague schlägt vor (basierend auf einer Reihe von Patienten, die in seiner Praxis untersucht wurden), eine Unterart der atopischen Dermatitis (AD) bei Patienten in Betracht zu ziehen, die diskoide und lichenoide Plaques aufweisen , die sich symmetrisch in Bereichen der Gliedmaßenstreckung befinden, unter Berücksichtigung der Beugungen , begleitet von Hyperkeratose. palmoplantar. Darüber hinaus ist Ig E im Serum sehr hoch , was sich klinisch in einem starken begleitenden Juckreiz niederschlägt . Die Histopathologie dieser Patienten weist auf psoriasiforme Veränderungen in der Epidermis und Eosinophile in der Dermis hin .
Um die diskoide und lichenoide AD vom Sulzberger-Garbe-Syndrom zu unterscheiden, wird diese Subvarietät als eigenständige Einheit vorgeschlagen.
| JAK-Inhibitoren bei atopischer Dermatitis Von Dr. Paula Luna (Argentinien). |
AD stellt eine sehr komplexe Physiopathogenese dar, die das Ergebnis einer Interaktion mehrerer Akteure ist. Es hat sicherlich eine genetische Grundlage, einen Pool von Genen, die mit der Hautbarriere, dem Juckreiz und der Immunität zusammenhängen. Andererseits ist es wichtig, die Rolle des Mikrobioms sowohl bei der Entstehung als auch bei der Dauer von AD hervorzuheben.
Heute ist bekannt, dass der Ursprung von AD nicht nur im Th2-Weg, sondern auch im Th1 liegt, wobei neben den allgemein bekannten Zytokinen 4 und 13 (hemmt durch Dupilumab) mehrere Zytokine beteiligt sind; IL 31 ist unter anderem für Pruritus verantwortlich.
In der klinischen Praxis beobachten wir unterschiedliche AD-Phänotypen, wobei unter anderem Unterschiede je nach Rasse, Evolutionszeit und Altersgruppe beobachtet werden. Dies könnte mit den jeweils beteiligten Entzündungswegen zusammenhängen. Die Behandlung dieser Pathologie entwickelt sich zu einem „Maßanzug“-Paradigma, bei dem die Behandlung für jeden Patienten auf der Grundlage des/der beteiligten Wegs/Wege angepasst wird. Aufgrund der großen klinischen und physiopathogenen Heterogenität gibt es vielfältige Therapiemöglichkeiten mit unterschiedlicher Wirksamkeit, von zellulären Inhibitoren bis hin zu hochspezifischen Inhibitoren wie biologischen.
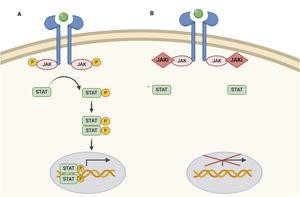
JAK-Inhibitoren sind mäßig spezifisch und hemmen mehrere Zytokine entlang des JAK-STAT-Signalwegs. Sie verhindern die Phosphorylierung und Aktivierung von STAT und damit dessen Wirkung auf der Ebene des Zellkerns, indem sie die genetische Transkription modulieren.
Dermosifiliographisches Protokoll vom 21. Januar 2021
JAK hat 4 Unterarten, die in Form von Dimeren funktionsfähig sind, als Rezeptor für verschiedene Zytokine fungieren und so physiologische Signalwege aktivieren.
Es gibt 3 JAK-Inhibitoren zur systemischen Anwendung: Baricitinib, Abrocitinib und Upadacitinib ; Das einzige in Europa für AD zugelassene (und in den USA im Zulassungsverfahren befindliche) ist Baricitinib, selektiv für JAK 1 und 2. Die anderen beiden sind selektiv für JAK 1. Baricitinib und Upadacitinib sind in unserem Land für die RA-Behandlung zugelassen. Derzeit laufen Forschungsprotokolle zur Zulassung von Baricitinib für AD bei Kindern und Erwachsenen; ebenso bei Upacitinib. Bei diesen drei Arzneimitteln handelt es sich um kleine Moleküle. Sie werden alle in einer einzigen täglichen Einnahme oral eingenommen und zeigen einen raschen Wirkungseintritt. Bisher konnte gezeigt werden, dass JAK-Inhibitoren bei Patienten mit AD, der Haupteinschränkung der Lebensqualität, eine große und sehr schnelle Linderung des Juckreizes bewirken.
Zu den unerwünschten Wirkungen dieser Klasse zählen Lymphome, Thrombosen und Infektionen, über die in Forschungsprotokollen nur minimal berichtet wurde. Baricitinib führt zu Eczema herpeticum und Herpes Zoster als Nebenwirkung; akneartige Upadacitinib-Reaktion, die häufig in Forschungsprotokollen beobachtet wird.
Diese Moleküle scheinen eine vielversprechende therapeutische Gruppe für Patienten mit AD zu sein.
| Update zur Behandlung kutaner Lymphome . |
| Update zur Behandlung von Mycosis fungoides Von Prof. Dr. José Sanches (Brasilien). |
Mycosis fungoides (MF) ist eine Neoplasie, die sich zu einer Vielzahl klinischer Manifestationen entwickelt. Anfänglich stellt es sich im Allgemeinen als kaum infiltrierte Plaques dar, die sich im Laufe der Zeit zu deutlich infiltrierten und ulzerierten Tumorläsionen entwickeln. Der Ursprung liegt in reifen, atypischen T-Lymphozyten, die zunächst die Epidermis befallen und mit fortschreitender Erkrankung auch die Lymphknoten, das periphere Blut und entfernte Organe befallen.
Bezüglich der Behandlung von MF gibt es keine klaren Leitlinien. Es handelt sich trotz der uns bisher zur Verfügung stehenden Behandlungen um eine unheilbare Krankheit.
Die Behandlung richtet sich nach dem Ausmaß der klinischen Manifestationen, der Art der vorhandenen Läsionen und dem Vorliegen einer systemischen Erkrankung. Wir haben topische und systemische Alternativen, die auf diesen zuvor genannten Parametern basieren. Die erste Gruppe umfasst: topische Kortikosteroide, Mechlorethamin, Carmustin, Bexaroten, Imiquimod und Resiquimode. Darüber hinaus sind als physikalische Behandlung sowohl die UVB- als auch die PUVA-Phototherapie, die photodynamische Therapie und die Strahlentherapie zu nennen.
Als Neuheit im Spektrum der topischen Behandlung gibt es Resiquimode . Es handelt sich um einen Toll-R 7/8-Agonisten, der im Anfangsstadium von MF sehr nützlich ist und eine sehr geringe Toxizität (lokale Reizung) aufweist. Es wird in Form eines Gels mit einer Konzentration von 0,03 oder 0,06 % angeboten. Rook et al. haben nachgewiesen, dass es bei 75 % der behandelten Patienten zu einer signifikanten Verbesserung kam, die behandelten Läsionen verbesserten sich alle und es war sogar eine Rückbildung der unbehandelten Läsionen zu beobachten.
Zu den systemischen Optionen gehören Medikamente, die die biologische Reaktion modifizieren, wie Interferon, Bexaroten, Acitretin, Alemtuzumab, Vorinostat/Romidepsin und Brentuximab Vedotin; Chemotherapeutika wie unter anderem Chlorambucil, Methotrexat, Gemcitabin und liposomales Doxorubicin. Diese letzten beiden sind die am häufigsten verwendeten Medikamente als Monotherapieoptionen. Auch extrakorporale Photopherese und Stammzelltransplantation sind Alternativen. Da es sich bei MF, wie oben erwähnt, derzeit um eine unheilbare Neubildung handelt und je nach Engagement im Einzelfall, erreichen die objektiven Ansprechraten bei diesen Behandlungen 30–50 %.
Im Frühstadium ist Interferon nach wie vor die erste therapeutische Wahl.
Brentuximab Vedotin , eine systemische Alternative, ist ein monoklonaler Antikörper wie Mogalizumab . Sie werden im Rahmen von Forschungsprotokollen untersucht, aber das erste würde offenbar eine Gesamtremissionsrate von 60 % aufweisen.
| Update zur Behandlung des Sezary-Syndroms von Dr. Paula Enz (Argentinien). |
Klinischer Verdacht im Zusammenhang mit Erythrodermie (Befall von mehr als 80 % der Körperoberfläche, T4), Lymphknoten und Sezary-Zellen im peripheren Blut.
> Aktuelles Konzept: Einsatz multimodaler, auf die Haut gerichteter Therapien in Verbindung mit rotierenden Tandem-Systemtherapien (mit neuen biologischen Wirkstoffen und Immunmodulatoren). Zytotoxische Chemotherapie nur im Zusammenhang mit einer Krankheitsprogression oder einem Versagen des klinischen Ansprechens auf eine andere etablierte Behandlung.
Die Behandlungen sind die zuvor genannten.
Die Phototherapie ist eine ausgezeichnete Alternative zur Behandlung von Pruritus, der bei diesen Patienten normalerweise einschränkend ist. Ebenso das Elektronenbad, das mit topischen Behandlungen, IFN oder Photopherese kombiniert werden kann. Mehr als 30 Gy sind wirksam, gehen jedoch mit einer erheblichen Hauttoxizität einher, oder bei lokalisierten Läsionen kann eine niedrigere Dosis angewendet werden. Dadurch bleibt die Möglichkeit, kürzere Behandlungen durchzuführen und nachzuarbeiten.
Die Photopherese ist eine gute erste Therapieoption. Es wird an zwei aufeinanderfolgenden Tagen alle 2, 3 oder 4 Wochen durchgeführt. Es kann allein oder in Kombination mit anderen Therapien angewendet werden. Das Gesamtansprechen liegt bei 40 %, die Studien umfassen jedoch nur wenige Patienten. Es erfordert ein „intaktes“ Immunsystem, daher wird die Anwendung vor einer immunsuppressiven Behandlung empfohlen.
Vorinostat und Romidepsin sind Inhibitoren der Histonacetylierung. Es gibt keine vergleichenden Studien zwischen den beiden. Beide haben eine Gesamtantwort von etwa 35 %.
Mogalizumab wurde 2018 zugelassen, es ist ein Anti-CCR4 . Bei SS liegt die Gesamtansprechrate bei 37 %. Es ist nützlich in Kombination mit anderen immunmodulatorischen Behandlungen.
Brentuximab wird bei Patienten angewendet , die >10 % CD30+-Lymphozyten in Hautbiopsien von Patienten mit SS aufweisen.
Alemtuzumab hat mehrere schwerwiegende unerwünschte Ereignisse (Immunsuppression und Infektionen mit Todesfolge). Für SS bei niedrigen Dosen werden Protokolle erstellt .
Bei Patienten mit gutem Allgemeinzustand, die auf die Erstbehandlung nicht angesprochen haben, ist die allogene Knochenmarktransplantation eine Alternative. Es kann eine heilende Alternative sein.
Monotherapie oder multimodale Behandlungen für SS? Viele Studien sprechen von der Kombination von Photopherese mit immunmodulatorischen Behandlungen, was zu einer Steigerung der Wirksamkeit mit einem Gesamtansprechen von nahezu 80 % führt. Eine multimodale Behandlung wäre die geeignete Vorgehensweise.
Enz bemerkte, dass ein Patient mit SS, der mit Photopherese und Bexaroten behandelt wurde, im Jahr 2020 zu einem ungünstigen Zeitpunkt seiner Hautbeteiligung eine COVID-Infektion hatte. Nach der Infektion entwickelte sich seine Haut sehr positiv.
> Zukunft: onkolytische Virotherapie mit Masern-Virusvektoren. Wird derzeit untersucht, hat aber bisher offenbar eine gute Resonanz.
| Behandlung primärer kutaner CD30-lymphoproliferativer Syndrome Von Dr. Mariana Arias (Argentinien). |
Dazu gehören das primär kutane anaplastische großzellige Lymphom (PLCLCL) und die lymphomatoide Papulose (LP). Sie machen etwa 25 % der primären kutanen Lymphome aus und sind nach MF und SS die zweithäufigste.
LACGCP: Erwachsene, Männer (3:1), 10-Jahres-Überlebensrate von 90 %. Erythematös-violette Tumoren mit schnellem Wachstum, die an jeder anatomischen Stelle im Allgemeinen groß ulzerieren können. In bis zu 40 % der Fälle kann es zu einer spontanen Rückbildung oder zu einem erneuten Auftreten kommen. Eine extrakutane Progression kommt häufig vor, insbesondere auf der Ebene der Lymphknoten, was die Prognose trübt. Ebenso verdienen Fälle von multiplen Tumoren in den unteren Extremitäten besondere Aufmerksamkeit; da sie in der Regel eine Lymphknotenbeteiligung aufweisen.
PL: Erwachsene, ausgezeichnete Prognose mit 100 % 10-Jahres-Überleben. Hohe Assoziation mit anderen Lymphomen, meist mit MF. Es verdient eine genaue Überwachung. Chronischer und wiederkehrender Ausschlag von Papeln und Knötchen, die ulzerieren können, am Rumpf und an den Gliedmaßen. Autoinvolution kommt sehr häufig vor und führt zu Schüben und Remissionen.
Es ist wichtig, eine Überbehandlung zu vermeiden, da diese Neoplasien eine sehr gute Prognose haben. Die Auswahl richtet sich nach dem Ausmaß und der Anzahl der Verletzungen. Wenig wissenschaftliche Belege für Fälle von Rezidiven oder systemischer Beteiligung.
Für LACGCP:
> Einzelne Läsion oder gruppierte Läsionen : chirurgische Entfernung in Verbindung mit lokaler Strahlentherapie oder lokalisierter Strahlentherapie nur mit einem seitlichen und tiefen Rand von 2 cm. Mit assoziierter Lymphknotenbeteiligung : lokalisierte Haut- und Lymphknoten-Strahlentherapie ± Brentuximab oder lokalisierte Haut- und Lymphknoten-Strahlentherapie (1. Linie NCCN 2021).
> Multifokale Läsionen : Brentuximab Vedotin als Erstlinientherapie (NCCN 2021); Als weitere Optionen wird eine Kombination aus systemischen Behandlungen +/- auf die Haut gerichteten Behandlungen (topisch oder physikalisch) vorgeschlagen.
Überprüfung von 10 Veröffentlichungen zu primären kutanen Lymphomen, die mit Brentuximab Vedotin behandelt wurden: 100 % der Patienten zeigten in einer mittleren Zeit von 5 Wochen ein vollständiges Ansprechen; Berichte über leichte Nebenwirkungen.
Bei LP kann eine Beobachtung durchgeführt werden, wenn man weiß, dass der limitierende Faktor das Vorhandensein eines zweiten assoziierten Lymphoms ist.
> Gruppierte Läsionen: Phototherapie oder topische Kortikosteroide.
> Erweiterte Läsionen : Methotrexat, Phototherapie (NCCN 2021), systemische Retinoide.
| Die biologische Revolution bei Psoriasis Von Prof. Dr. Jonathan Barker (Vereinigtes Königreich). |
In den letzten 30 Jahren hat sich das Verständnis der Psoriasis unglaublich weiterentwickelt. In den 80er Jahren betrachteten wir ihre Entstehung als eine Störung der epidermalen Differenzierung, weshalb man begann, immunsuppressive Medikamente wie Methotrexat einzusetzen. Dann wandte man sich der Immunologie zu und begann, hier Ciclosporin einzusetzen, basierend auf einer Schweizer Studie, in der Patienten mit rheumatoider Arthritis untersucht wurden, die mit diesem Medikament behandelt wurden. In den letzten 20 Jahren haben wir uns zum Ziel gesetzt, jeden einzelnen Fall zu stratifizieren, um eine gezielte Behandlung anbieten zu können. Dabei haben wir verstanden, dass kutane Lymphozyten eine große Anzahl von Zytokinen synthetisieren, die ein komplexes Netzwerk bilden, das zu unterschiedlichen klinischen Zuständen führt. Wir befinden uns im Zeitalter der biologischen Behandlungen. Sie haben zweifellos den Verlauf der Psoriasis verändert, aber wie jede Behandlung haben sie ihre Grenzen. Sie sind sehr teuer, sie sind nicht bei allen Arten von Psoriasis wirksam, ihre Wirkung lässt mit der Zeit nach, natürlich haben sie ihre Nebenwirkungen usw.
Im Vereinigten Königreich sind biologische Behandlungen vom National Health Service mit strengen Kontrollen zugelassen. Die Behandlungsteams messen die Konzentrationen der verschiedenen biologischen Arzneimittel einen Monat nach Beginn der Behandlung, da sich herausstellte, dass diejenigen Patienten, die einen bestimmten gewünschten Konzentrationsbereich derselben im Serum nicht erreichen, Kandidaten dafür sind, die biologischen Arzneimittel auszusetzen und die Behandlung durch eine andere zu ersetzen . . Sie tun dies als Strategie, um Ressourcen zu sparen, da sie wissen, dass diese Patienten aufgrund eines biologischen Wirkstoffs anfälliger für primäre Ausfälle sind. Barker sagte, sie hätten Adalimumab pünktlich durchgeführt.
Es ist unmöglich, die COVID-Pandemie im Zusammenhang mit diesen Patienten nicht zu erwähnen. Barker führte Aufzeichnungen über die Nachbeobachtung von Patienten mit Psoriasis durch, die mit Biologika behandelt wurden, daher werden uns diese Ergebnisse in Zukunft vorliegen. Was den COVID-Impfstoff betrifft, ist nicht bekannt, wie groß die Schutzwirkung bei dieser Patientengruppe sein wird und wie sicher er ist. Während man mit Empfehlungen vorsichtig sein muss, empfiehlt er es seinen Patienten.
Barker schätzt, dass in den nächsten zehn Jahren verschiedene topische Optionen auftauchen und der Einsatz von Biomarkern in der klinischen Praxis zunehmen wird, um Patienten zu kategorisieren und „maßgeschneiderte“ gezielte Behandlungen anzubieten.
| Neue Diagnosemethoden bei Lepra Von Dr. Heitor Goncalves (Brasilien). |
Es werden diagnostische Methoden vorgeschlagen, um verschiedene diagnostische Herausforderungen der klinischen Praxis einfacher zu charakterisieren: unbestimmte Lepra ohne Änderung der Empfindlichkeit, Differenzialdiagnose zwischen Rückreaktion und Rezidiv bei paucibacillären Patienten, Differenzialdiagnose zwischen Rückreaktion und Rezidiv bei multibazillären Patienten, rein neuronale Formen, Silent Neural Lähmung und Frühdiagnose von multi- und paucibazillären Patienten in Endemiegebieten.
> Diagnosetools:
Klinisch DAS WICHTIGSTE (thermische Empfindlichkeit, motorische Beeinträchtigung);
• Labor: Baciloskopie (besonders wichtig bei multibakteriellen Patienten).
• Histopathologie (Differenzialdiagnose von Rezidiven oder Reaktionsmustern bei MB).
• Histamintest (Differentialdiagnose unbestimmter Formen, nützlich bei pädiatrischen Patienten zur Unterscheidung von akromianten Ekzematiden, die bei AD beobachtet werden).
• Elektromyogramm: Bis zu 40 % der Patienten, die noch keine klinischen Manifestationen einer Neuritis aufweisen, weisen zuvor eine EMG-Beteiligung auf; dd mit anderen Pathologien, die Neuropathien verursachen; Der Nachteil ist, dass es keine pathognomonischen Veränderungen hervorruft.
• Neuronaler Ultraschall : nützlich zur Beurteilung der Verdickung, insbesondere in Fällen, in denen eine Dekompression erforderlich ist, und zur Durchführung gezielter Biopsien.
• Serologie : Es werden Anti-Lepra-Antikörper gemessen . Je höher die Ac-Konzentration des Patienten ist, desto größer ist die humorale Reaktion und daher wird von einem multibakteriellen Patienten ausgegangen. Nützlich für die Überwachung von Patienten, die in Endemiegebieten leben. Eine A+-Serologie bedeutet nicht, dass der Patient krank ist, sondern vielmehr, dass er Kontakt mit dem Bazillus M. leprae hatte und daher eine engmaschige Nachsorge erforderlich ist. Der Nachweis erfolgt mit der ELISA-Methode mit einer Spezifität von 98 %. Für paucibacilläre Patienten von geringem Wert.
• M. leprae- PCR : Erkennt lebende oder lebensfähige Bazillus, nützlich bei neuronalen, unbestimmten Formen, zur Unterscheidung von Reaktionsphänomenen einer erneuten Infektion (es kann sich um eine andere Art von Bazillus handeln) oder eines Wiederauftretens (derselbe Bazillus) und für paucibacilläre Patienten.
- Konventioneller Typ: verstärkt sowohl lebende als auch tote Bakterien.
- Quantitativer Typ in Echtzeit.
- Reverse Transkriptase: +; bedeutet lebender Bazillus (Rezidiv und keine Reaktion).
• Nervenbiopsie: sensorischer Nerv; selten auf motorische Nerven (hohe Verletzungsgefahr). Richten Sie es mit Ultraschall aus. PCR kann am Nerv durchgeführt werden.
• Untersuchung des Bazillus-Genoms: nützlich für Populationsstudien, um Rückfall-Reinfektionen zu bestimmen. Die Möglichkeit der Entwicklung eines Impfstoffs wird geprüft.















