| Farmacodermie Di Ana Clara Torre (Argentina). |
Qualsiasi effetto dannoso o indesiderato che si manifesta dopo l’uso di un farmaco a una dose profilattica, diagnostica o terapeutica. L’OMS stima che rappresenti circa il 15% di tutte le reazioni avverse ai farmaci.
Si possono classificare in semplici o non complicate (90-95% dei casi), in cui si osserva un lieve coinvolgimento cutaneo, senza coinvolgimento sistemico, e complesse o complicate (2-5% dei casi), che presentano coinvolgimento. grave coinvolgimento cutaneo e/o sistemico. In quest’ultimo gruppo si osservano solitamente alterazioni di laboratorio, segno di Nikolsky, pustole generalizzate, lesioni a bersaglio, lesioni purpuriche o eritroderma.
In base alla gravità della condizione, possono essere affrontati nel modo seguente:
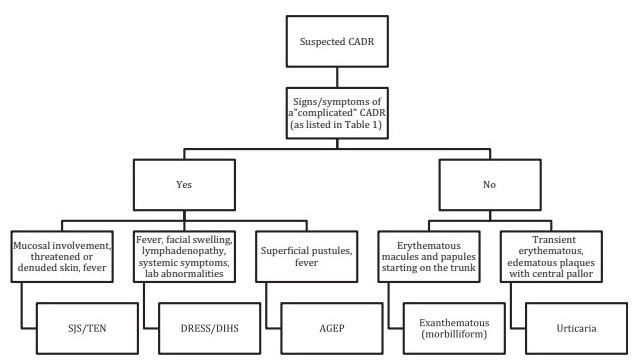
CADR: reazione avversa cutanea al farmaco
AGEP: rash pustoloso acuto generalizzato
SJS/TEN: sindrome di Steven-Johnson/necrolisi epidermica tossica
DRESS/DIHS: reazione avversa al farmaco con eosinofilia e sintomi sistemici/sindrome da ipersensibilità indotta da farmaci.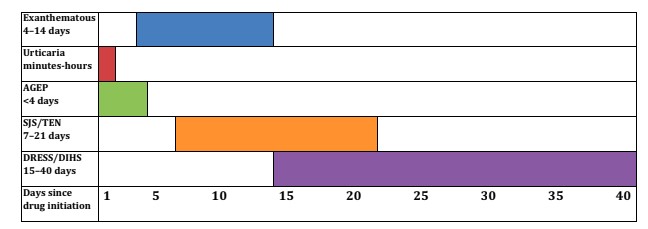
Va notato che non tutti i sintomi presentano lo stesso tempo di latenza dopo l’introduzione del farmaco, costringendoci a mettere in discussione anche quelli consumati fino a 40 giorni prima della comparsa della patologia cutanea.
Swanson L, Colven RM. Approccio al paziente con sospetta reazione cutanea avversa al farmaco. Med Clin Nord Am. 2015 novembre;99(6):1337-48
Per quanto riguarda lo studio di queste condizioni cliniche, è essenziale uno studio di laboratorio, sia per valutare il danno d’organo bersaglio, sia per escludere altre diagnosi differenziali.
| Mastocitosi infantile A cura del Prof. Dott. Antonio Torrelo (Spagna). |
Insieme di malattie maligne in cui si osserva un accumulo patologico di mastociti in diversi tessuti. Sono dovuti ad una mutazione clonale del gene kit. Può iniziare in età diverse e presentare fenotipi clinici diversi, il che rende la sua classificazione più complessa.
L’OMS riconosce solo 3 sottotipi nei pazienti pediatrici in base al numero di lesioni, tralasciando alcuni parametri come la genetica, la morfologia, l’istologia o il comportamento della malattia. Questa classificazione genera molte polemiche, utilizzando termini ambigui, spesso non riuscendo a classificare i pazienti che valutiamo quotidianamente.
Torrelo, vista la sua esperienza su questo argomento, propone 2 tipi di mastocitosi infantile , tipo 1 e 2. Il primo, o classico, è il tipo osservato negli adulti. Tipo 2 o ben differenziato, frequentemente osservato nei bambini.
Tipo 1 : piccole lesioni cutanee melaniche maculari o papulari; numero variabile di infortuni. La dermatoscopia evidenzia teleangectasie. Segno più audace ma difficile da provocare. Occasionalmente si possono vedere delle vesciche. L’istopatologia mostra mastociti perivascolari del fuso in numero moderato. Raramente è accompagnato da sintomi sistemici. Mastociti con mutazione del gene kit nel codone 815 o 816. I mastociti del midollo osseo sono a forma di fuso e marcano positivamente CD2 e CD25 nella citometria a flusso. Non risponde all’imatinib ma risponde alla midostaurina.
È la varietà più osservata durante l’infanzia.
Tipo 2 : macule, placche, papule, noduli di grandi dimensioni e possono essere presenti un gran numero di lesioni. Spesso si possono osservare vescicole. Con l’evoluzione si possono osservare forme anetodermiche ed eritrodermiche. Segno più Darier + prominente. L’istopatologia mostra mastociti rotondi e granulati, con nuclei polilobulati. I sintomi sistemici sono comuni. I mastociti del midollo osseo segnano negativamente alla citometria a flusso per CD2 e CD25. Le mutazioni nel kit non si osservano nel codone 815 o 816, risponde molto bene all’inibitore della tirosina chinasi imatinib. Questa varietà corrisponde al 5-10% dei casi di mastocitosi osservati nella vita adulta, poiché tende a scomparire.
| Dermatite atopica discoide e lichenoide A cura del Prof. Dr. José Ollague (Ecuador). |
Ollague propone (sulla base di una serie di pazienti valutati nel suo studio) di considerare una sottovarietà di dermatite atopica (AD) in pazienti che presentano placche discoidi e lichenoidi , localizzate simmetricamente nelle aree di estensione degli arti, rispettando le flessioni , accompagnate da ipercheratosi. palmo-plantare. Inoltre, le Ig E sono molto elevate nel siero, il che si traduce clinicamente in un intenso prurito accompagnatorio . L’istopatologia di questi pazienti evidenzia la presenza di alterazioni psoriasiformi nell’epidermide e di eosinofili nel derma .
Per differenziare l’AD discoide e lichenoide dalla sindrome di Sulzberger-Garbe, questa sottovarietà viene proposta come entità indipendente.
| Inibitori JAK nella dermatite atopica Della Dott.ssa Paula Luna (Argentina). |
L’AD presenta una fisiopatogenesi molto complessa, frutto di un’interazione di più attori. Certamente ha una base genetica, un pool di geni, legati alla barriera cutanea, al prurito e all’immunità. D’altro canto è importante evidenziare il ruolo del microbioma sia nella genesi che nella perennità dell’AD.
Oggi è noto che l’origine dell’AD non risiede esclusivamente nella via Th2 ma anche in quella Th1, con molteplici citochine coinvolte oltre alle ampiamente conosciute 4 e 13 (inibite da dupilumab); IL 31 responsabile, tra gli altri, del prurito.
Nella pratica clinica osserviamo diversi fenotipi di AD, con differenze osservate a seconda della razza, del tempo evolutivo e della fascia di età, tra gli altri fattori in questione. Ciò potrebbe corrispondere alle vie infiammatorie coinvolte in ciascun caso particolare. Il trattamento di questa patologia si sta evolvendo verso un paradigma “su misura”, abbinando il trattamento a ciascun paziente in base al/ai percorso/i coinvolto/i. La grande eterogeneità clinica e fisiopatogena fa sì che esistano molteplici opzioni terapeutiche, di diversa efficacia, dagli inibitori cellulari agli inibitori altamente specifici come quelli biologici.
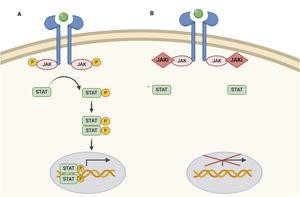
Gli inibitori JAK sono moderatamente specifici, inibendo diverse citochine lungo la via JAK-STAT. Impediscono la fosforilazione, l’attivazione e quindi la sua azione di STAT a livello del nucleo cellulare modulando la trascrizione genetica.
Verbale dermosifiliografico del 21 gennaio 2021
JAK ha 4 sottovarietà, essendo funzionali sotto forma di dimeri, agendo come recettore per diverse citochine e attivando così percorsi fisiologici.
Esistono 3 inibitori JAK per uso sistemico: baricitinib, abrocitinib e upadacitinib ; L’unico approvato in Europa per l’AD (e in fase di approvazione negli USA) è baricitinib, selettivo per JAK 1 e 2. Gli altri due sono selettivi per JAK 1. Baricitinib e upadacitinib sono approvati nel nostro Paese per il trattamento dell’artrite reumatoide. Sono attualmente in corso protocolli di ricerca per approvare baricitinib per l’AD nei bambini e negli adulti; allo stesso modo con upacitinib. Questi 3 farmaci sono considerati piccole molecole, tutti vengono ingeriti per via orale in un’unica assunzione giornaliera, presentando una rapida insorgenza d’azione. Finora si è visto che gli inibitori della JAK forniscono un grande sollievo dal prurito e molto rapidamente nei pazienti con AD, la principale limitazione della qualità della vita.
Tra gli effetti avversi di classe possiamo citare linfomi, trombosi e infezioni, che sono state minimamente riportate nei protocolli di ricerca. Baricitinib presenta l’eczema erpetico e l’herpes zoster come effetto avverso; reazione acneiforme di upadacitinib, comunemente osservata nei protocolli di ricerca.
Queste molecole sembrerebbero essere un gruppo terapeutico promettente per i pazienti affetti da AD.
| Aggiornamento sul trattamento dei linfomi cutanei . |
| Aggiornamento sul trattamento della micosi fungoide A cura del Prof. Dr. José Sanches (Brasile). |
La micosi fungoide (MF) è una neoplasia che evolve in un’ampia gamma di manifestazioni cliniche. Inizialmente si presenta generalmente come placche appena infiltrate, che evolvono nel tempo in lesioni tumorali notevolmente infiltrate e ulcerate. La sua genesi risiede nei linfociti T maturi e atipici, che inizialmente coinvolgono l’epidermide e con il progredire della malattia colpiscono anche i linfonodi, il sangue periferico e gli organi distanti.
Per quanto riguarda il trattamento della MF, non esistono linee guida chiare. È una patologia incurabile nonostante le cure di cui disponiamo ad oggi.
Il trattamento si basa sull’entità delle manifestazioni cliniche, sul tipo di lesioni presenti e sulla presenza di malattia sistemica. Disponiamo di alternative topiche e sistemiche basate su questi parametri precedentemente menzionati. Il primo gruppo comprende: corticosteroidi topici, mecloretamina, carmustina, bexarotene, imiquimod e resiquimode. Inoltre, sia la fototerapia UVB che quella PUVA, la terapia fotodinamica e la radioterapia possono essere menzionate come trattamento fisico.
Come novità nello spettro dei trattamenti topici c’è il resiquimode . È un agonista Toll-R 7/8 molto utile nelle fasi iniziali della MF e presenta una tossicità molto bassa (irritazione locale). Si presenta sotto forma di gel, allo 0,03 o allo 0,06%. Rook et al hanno dimostrato che si ha un miglioramento significativo nel 75% dei pazienti trattati, tutte le lesioni trattate migliorano ed è stata osservata anche la regressione delle lesioni non trattate.
Tra le opzioni sistemiche figurano i farmaci modificanti la risposta biologica come interferone, bexarotene, acitretina, alemtuzumab, vorinostato/romidepsina e brentuximab vedotin; agenti chemioterapici, come clorambucile, metotrexato, gemcitabina, doxorubicina liposomiale, tra gli altri. Questi ultimi due sono i farmaci più utilizzati come opzioni di monoterapia. Altre alternative sono la fotoferesi extracorporea e il trapianto di cellule staminali. Come accennato in precedenza, dato che la MF è attualmente una neoplasia incurabile e, a seconda dell’impegno di ciascun caso particolare, i tassi di risposta obiettiva con questi trattamenti raggiungono il 30-50%.
Per gli stadi iniziali, l’interferone continua ad essere la prima scelta terapeutica.
Brentuximab vedotin , un’alternativa sistemica, è un anticorpo monoclonale come mogalizumab . Sono allo studio nell’ambito di protocolli di ricerca, ma i primi avrebbero apparentemente un tasso di remissione complessiva del 60%.
| Aggiornamento sul trattamento della sindrome di Sezary A cura della Dott.ssa Paula Enz (Argentina). |
Sospetto clinico nel contesto di eritroderma (coinvolgimento di oltre l’80% della superficie corporea, T4), linfonodi e cellule di Sezary nel sangue periferico.
> Concezione attuale: utilizzo di terapie multimodali dirette alla pelle associate a terapie sistemiche tandem rotanti (con nuovi agenti biologici e immunomodulatori). Chemioterapia citotossica solo in un contesto di progressione della malattia o di fallimento nella risposta clinica ad un altro trattamento stabilito.
Le cure sono quelle citate in precedenza.
La fototerapia è un’ottima alternativa per trattare il prurito, che solitamente è limitante in questi pazienti. Allo stesso modo, il bagno di elettroni, che può essere combinato con trattamenti topici, IFN o fotoferesi. Più di 30 Gy sono efficaci, ma sono associati a una significativa tossicità cutanea, oppure è possibile utilizzare una dose inferiore in lesioni localizzate. Ciò lascia la possibilità di eseguire trattamenti più brevi e di ritracciare.
La fotoferesi è una buona prima opzione terapeutica. Si effettua 2 giorni consecutivi ogni 2, 3 o 4 settimane. Può essere utilizzato da solo o in combinazione con altre terapie. La risposta complessiva è del 40%, ma gli studi riguardano pochi pazienti. Richiede un sistema immunitario “intatto”, quindi si consiglia di utilizzarlo prima del trattamento immunosoppressivo.
Vorinostat e romidepsin sono inibitori dell’acetilazione degli istoni. Non esistono studi comparativi tra i due. Entrambi hanno risposte complessive di circa il 35%.
Mogalizumab è stato approvato nel 2018, è un anti-CCR4 . Ha una risposta complessiva del 37% nella SS. È utile in combinazione con altri trattamenti immunomodulatori.
Brentuximab è utilizzato nei pazienti che hanno >10% di linfociti CD30+ nelle biopsie cutanee di pazienti con SS.
Alemtuzumab presenta diversi eventi avversi gravi (immunosoppressione e infezioni con morte), si stanno elaborando protocolli per la SS a basse dosi .
Il trapianto allogenico di midollo osseo è un’alternativa nei pazienti con buone condizioni generali che non hanno risposto al trattamento di prima linea. Può essere un’alternativa curativa.
Trattamenti monoterapia o multimodali per la SS? Molti studi parlano di abbinare la fotoferesi ai trattamenti immunomodulatori, che ha un aumento di efficacia con una risposta complessiva vicina all’80%. Il trattamento multimodale sarebbe la linea d’azione appropriata.
Enz ha commentato che un paziente con SS in trattamento con fotoferesi e bexarotene ha avuto un’infezione da COVID nel corso del 2020 in un brutto momento per quanto riguarda il coinvolgimento della sua pelle. Dopo l’infezione, la sua pelle si è evoluta in modo molto favorevole.
> Futuro: viroterapia oncolitica, con vettori virali del morbillo. In fase di studio, ma a quanto pare finora ha avuto una buona risposta.
| Trattamento delle sindromi linfoproliferative cutanee primarie CD30 A cura della Dott.ssa Mariana Arias (Argentina). |
Comprende il linfoma cutaneo anaplastico primario a grandi cellule (PLCLCL) e la papulosi linfomatoide (LP). Questi rappresentano circa il 25% dei linfomi cutanei primitivi, secondi per frequenza dopo MF e SS.
LACGCP: adulti, uomini (3:1), sopravvivenza a 10 anni del 90%. Tumori eritemato-violacei a crescita rapida, che possono ulcerarsi, generalmente di grandi dimensioni, in qualsiasi sede anatomica. Fino al 40% dei casi può avere una regressione spontanea o recidivare. La progressione extracutanea è comune, soprattutto a livello linfonodale, e ciò offusca la prognosi. Allo stesso modo, meritano un’attenzione particolare i casi di tumori multipli agli arti inferiori; poiché di solito presentano un coinvolgimento linfonodale.
PL: adulti, prognosi eccellente con sopravvivenza a 10 anni del 100%. Alta associazione con altri linfomi, solitamente con MF. Merita un attento monitoraggio. Eruzione cronica e ricorrente di papule e noduli che possono ulcerarsi, sul tronco e sugli arti. L’autoinvoluzione è molto comune e provoca episodi di riacutizzazioni e remissioni.
È fondamentale evitare trattamenti eccessivi poiché queste neoplasie hanno una prognosi molto buona. La scelta si basa sull’entità e sul numero degli infortuni. Poche prove scientifiche per casi di recidiva o coinvolgimento sistemico.
Per LACGCP:
> Lesione singola o lesioni raggruppate : asportazione chirurgica associata a radioterapia localizzata o radioterapia solo localizzata con margine sia lateralmente che in profondità di 2 cm. Con coinvolgimento linfonodale associato : radioterapia localizzata della pelle e dei linfonodi ± brentuximab o radioterapia localizzata della pelle e dei linfonodi (1a linea NCCN 2021).
> Lesioni multifocali : brentuximab vedotin come 1a linea (NCCN 2021); Come altre opzioni, viene proposta un’associazione di trattamenti sistemici +/- trattamenti diretti sulla pelle (topici o fisici).
Revisione di 10 pubblicazioni su linfomi cutanei primitivi trattati con brentuximab vedotin: il 100% dei pazienti ha presentato una risposta completa in un tempo mediano di 5 settimane; segnalazioni di lievi effetti avversi.
Per l’LP l’osservazione può essere fatta sapendo che il fattore limitante è la presenza di un secondo linfoma associato.
> Lesioni raggruppate: fototerapia o corticosteroidi topici.
> Lesioni estese : metotrexato, fototerapia (NCCN 2021), retinoidi sistemici.
| La rivoluzione biologica nella psoriasi A cura del Prof. Dr. Jonathan Barker (Regno Unito). |
Negli ultimi 30 anni, la comprensione della psoriasi si è evoluta incredibilmente. Negli anni ’80 si considerava la sua genesi come un disordine della differenziazione epidermica, per questo si cominciarono ad utilizzare farmaci immunosoppressori come il metotressato. Poi l’attenzione si è spostata sull’immunologia e qui si è iniziato ad utilizzare la ciclosporina, sulla base di uno studio svizzero che ha valutato pazienti affetti da artrite reumatoide trattati con questo farmaco. Negli ultimi 20 anni abbiamo mirato a stratificare ogni caso particolare per offrire un trattamento mirato, comprendendo che i linfociti cutanei sintetizzano un gran numero di citochine che compongono una rete intricata che dà origine a diverse condizioni cliniche. Ci troviamo nell’era delle cure biologiche. Hanno indubbiamente cambiato il decorso della psoriasi, ma come ogni trattamento hanno i loro limiti. Sono molto costosi, non sono efficaci in tutti i tipi di psoriasi, il loro effetto diminuisce con il tempo, ovviamente hanno i loro effetti negativi, ecc.
Nel Regno Unito i trattamenti biologici sono approvati dal Servizio Sanitario Nazionale con severi controlli. Le équipe curanti misurano i livelli dei diversi farmaci biologici un mese dopo l’inizio del trattamento poiché si è visto che quei pazienti che non raggiungono un certo intervallo di concentrazione desiderata degli stessi nel siero sono candidati a sospendere il biologico e ruotare il trattamento per un altro . . Lo fanno come strategia per risparmiare risorse sapendo che questi pazienti sono più inclini a fallimenti primari dovuti a un agente biologico. Barker ha detto che hanno effettuato adalimumab in tempo.
Impossibile non menzionare la pandemia COVID in relazione a questi pazienti. Barker ha effettuato delle registrazioni sul follow-up dei pazienti affetti da psoriasi trattati con farmaci biologici, quindi avremo questi risultati in futuro. Per quanto riguarda il vaccino anti-COVID, non si sa quanto effetto protettivo avrà in questo gruppo di pazienti e quanto sarà sicuro. Anche se devi essere cauto con le raccomandazioni, lo raccomanda ai suoi pazienti.
Barker stima che nei prossimi 10 anni emergeranno diverse opzioni topiche e si amplierà l’uso dei biomarcatori nella pratica clinica, per categorizzare i pazienti e offrire trattamenti mirati “su misura”.
| Nuovi metodi diagnostici nella lebbra A cura del Dr. Heitor Goncalves (Brasile). |
Propone metodi diagnostici per caratterizzare più facilmente diverse sfide diagnostiche della pratica clinica: lebbra indeterminata senza alterazione della sensibilità, diagnosi differenziale tra reazione inversa e recidiva nei pazienti paucibacillari, diagnosi differenziale tra reazione inversa e recidiva nei pazienti multibacillari, forme neurali pure, Silent neurale paralisi e diagnosi precoce di pazienti multi- e paucibacillari in aree endemiche.
> Strumenti diagnostici:
Clinico IL PIÙ IMPORTANTE (sensibilità termica, compromissione motoria);
• Laboratorio: Baciloscopia (particolarmente importante nei pazienti multibacillari).
• Istopatologia (diagnosi differenziale di recidiva o pattern reattivo nel MB).
• Test dell’istamina (diagnosi differenziale delle forme indeterminate, utile nei pazienti pediatrici per distinguerla dalle eczematidi acromianti osservate nell’AD).
• Elettromiogramma: fino al 40% dei pazienti che non presentano ancora manifestazioni cliniche di neurite presentano preventivamente un coinvolgimento dell’EMG; dd con altre patologie che causano neuropatie; Lo svantaggio è che non dà cambiamenti patognomonici.
• Ecografia neurale : utile per valutare l’ispessimento, soprattutto nei casi in cui è necessaria la decompressione e per eseguire biopsie mirate.
• Sierologia : si misurano gli anticorpi anti-lebbra . Maggiore è la concentrazione di Ac del paziente, si deduce una maggiore risposta umorale e, quindi, viene interpretato un paziente multibacillare. Utile per monitorare i pazienti che vivono in aree endemiche. Una sierologia A+ non implica che il paziente sia malato, ma piuttosto che abbia avuto un contatto con il bacillo di M. leprae e, pertanto, richiede un attento follow-up. Il rilevamento viene effettuato con il metodo ELISA, con una specificità del 98%. Di scarso valore per i pazienti paucibacillari.
• M. leprae PCR : rileva bacilli vivi o vitali, utile per le forme neurali, indeterminate, per differenziare fenomeni reattivi di reinfezione (può essere un altro tipo di bacillo) o di recidiva (stesso bacillo) e per pazienti paucibacillari.
- Tipo convenzionale: amplifica sia i bacilli vivi che quelli morti.
- Tipo quantitativo in tempo reale.
- Trascrittasi inversa: +; significa bacillo vivo (recidiva e non reazione).
• Biopsia del nervo: nervo sensoriale; raramente sui nervi motori (alto rischio di lesioni). Dirigerlo con l’ecografia. La PCR può essere eseguita sul nervo.
• Studio del genoma del bacillo: utile per studi di popolazione, per determinare recidive di reinfezione. È allo studio la possibilità di sviluppare un vaccino.















