Strukturierte Zusammenfassung
Einführung
Neuropsychiatrische Störungen und psychische Erkrankungen sind die Hauptursache für die Krankheitslast in den Vereinigten Staaten. Zehntausende Sequenzvarianten im menschlichen Genom wurden mit der Ätiologie dieser Erkrankungen in Verbindung gebracht. Die Aufklärung der Rolle der identifizierten Risikovarianten bleibt jedoch eine Herausforderung, da sich die meisten von ihnen außerhalb proteinkodierender Regionen befinden und derzeit keine funktionelle Annotation aufweisen. Diese Krankheitsrisikovarianten üben wahrscheinlich ihren Einfluss aus, indem sie transkriptionelle Regulierungselemente stören und dadurch die Genexpression in Zelltypen modulieren, die für neuropsychiatrische Störungen relevant sind. Jüngste Fortschritte in der Einzelzelltechnologie haben ein hohes Maß an zellulärer Heterogenität im gesamten menschlichen Gehirn offenbart. Allerdings sind die Transkriptionsregulationssequenzen, die die Identität und Funktion jedes einzelnen Gehirnzelltyps steuern, noch nicht abgegrenzt, was unsere Fähigkeit zur Interpretation nicht-kodierender Krankheitsrisikovarianten beeinträchtigt.
Grundlegender Grund
Herkömmlicherweise können Transkriptionsregulationssequenzen durch den Nachweis der Chromatinzugänglichkeit bestimmt werden, die typischerweise mit der Transkriptionsfaktorbindung und der Chromatinumgestaltung einhergeht. Allerdings fehlen in früheren Katalogen transkriptionsregulierender Elemente Informationen über die zelltypspezifischen Aktivitäten jedes Elements, da große Gewebeproben verwendet werden. Jüngste technologische Fortschritte haben es uns ermöglicht, die Zugänglichkeit von Chromatin auf Einzelzellebene zu analysieren und so zelltypspezifische Karten transkriptioneller Regulierungselemente für komplexe Organe wie das menschliche Gehirn zu erstellen.
Ergebnisse
In dieser Studie präsentieren wir eine umfassende Analyse der Zugänglichkeit von Chromatin im menschlichen Gehirn auf Einzelzellebene und umfassen eine Sammlung von 1,1 Millionen Zellen aus 42 verschiedenen Gehirnregionen bei drei neurotypischen erwachsenen Probanden. Wir haben diesen Chromatinatlas verwendet, um 107 verschiedene Gehirnzelltypen zu definieren und den Chromatin-Zugänglichkeitsstatus von 544.735 mutmaßlichen Transkriptionsregulationselementen in diesen Zelltypen zu ermitteln.
Eine beträchtliche Anzahl dieser regulatorischen Elemente zeigte auch in Gehirnzellen von Mäusen Sequenzkonservierung und Chromatinzugänglichkeit , was ihre funktionelle Bedeutung unterstreicht. Durch integrative Analyse haben wir viele mutmaßliche Transkriptionsregulationselemente mit potenziellen Zielgenen verknüpft. Darüber hinaus haben wir diesen Atlas genutzt, um Zelltypen vorherzusagen, die für 19 neuropsychiatrische Merkmale und Störungen relevant sind. Schließlich haben wir Modelle für maschinelles Lernen entwickelt, um die regulatorische Funktion von Krankheitsrisikovarianten vorherzusagen. Wir haben diesen Atlas über das interaktive Webportal CATLAS (www.catlas.org) der Öffentlichkeit kostenlos zur Verfügung gestellt.
Schlussfolgerungen Der Einzelzell-Chromatinatlas des menschlichen Gehirns stellt eine wertvolle Ressource für die neurowissenschaftliche Gemeinschaft dar. Es bietet Einblicke in die Genregulationsprogramme, die die Vielfalt der Gehirnzelltypen prägen, und hilft bei der Interpretation der funktionellen Rollen von Krankheitsrisikovarianten, die außerhalb proteinkodierender Regionen liegen. Dieser Atlas verspricht in Kombination mit anderen molekularen und anatomischen Daten, unser Verständnis der Gehirnfunktion und Neuropathologie zu verbessern und bietet letztendlich Möglichkeiten für wirksamere Ansätze zur Behandlung neuropsychiatrischer Störungen . |
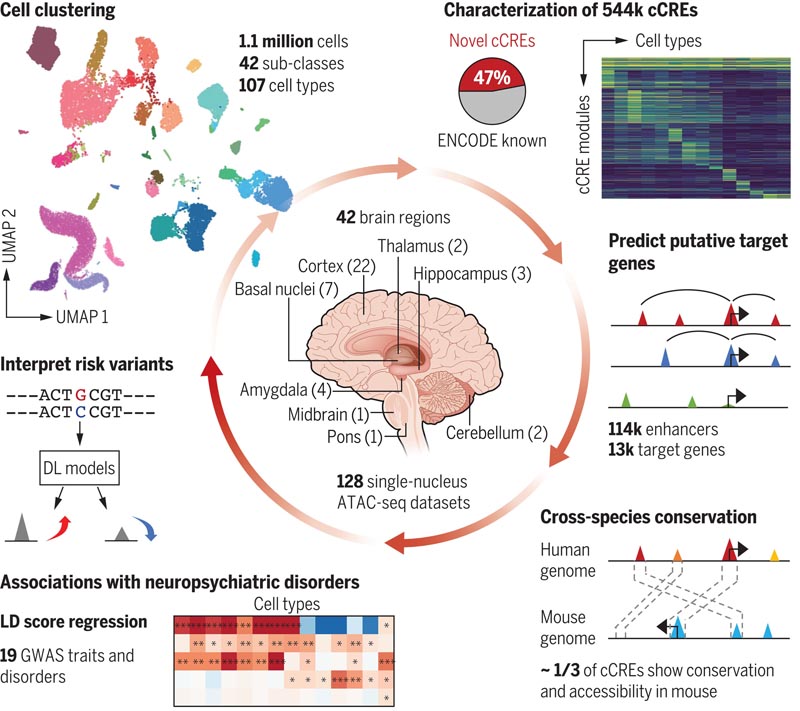
Abbildung: Einzelzellanalyse der Zugänglichkeit von Chromatin im menschlichen Gehirn . Kandidaten für cis-regulatorische Elemente (cCREs), die für verschiedene menschliche Gehirnzelltypen spezifisch sind, wurden mithilfe eines Single-Core-Assays für Transposase-zugängliches Chromatin durch Sequenzierung (snATAC-seq) identifiziert und durch integrative Analyse mit mutmaßlichen Zielgenen verknüpft. Der Einsatz von cCRE wurde genutzt, um Gehirnzelltypen vorherzusagen, die für neuropsychiatrische Merkmale und Störungen relevant sind, und um Modelle für maschinelles Lernen zu trainieren, um die Funktion nicht-kodierender Risikovarianten zu interpretieren. UMAP, Multiple Uniform Approach and Projection; DL, tiefes Lernen; LD, Bindungsungleichgewicht; GWAS, genomweite Assoziationsstudie.
Kommentare
Forscher kartieren genetische Schalter und Gehirnzelltypen, die mit Schizophrenie, bipolarer Störung, Alzheimer-Krankheit und schwerer Depression verbunden sind.
In einer großen multiinstitutionellen Initiative unter der Leitung der University of California, San Diego, analysierten Forscher mehr als eine Million menschliche Gehirnzellen, um detaillierte Karten genetischer Schalter in Gehirnzelltypen zu erstellen und deckten Zusammenhänge zwischen bestimmten Zelltypen und mehreren Faktoren auf. häufig bei neuropsychiatrischen Erkrankungen. Das Team entwickelte außerdem Werkzeuge der künstlichen Intelligenz, um den Einfluss einzelner Hochrisiko-Genvarianten auf diese Zellen und ihren möglichen Beitrag zur Krankheit vorherzusagen.
Die neue Arbeit, die in einer Sonderausgabe von Science veröffentlicht wurde, ist Teil der Brain Research through Advancing Innovative Neurotechnologies Initiative (BRAIN Initiative) der National Institutes of Health, die 2014 ins Leben gerufen wurde. Die Initiative zielt darauf ab, das Verständnis des Gehirns von Säugetieren zu revolutionieren Teilweise durch die Entwicklung neuer Neurotechnologien zur Charakterisierung neuronaler Zelltypen.
Jede menschliche Gehirnzelle enthält die gleiche DNA-Sequenz, aber verschiedene Zelltypen nutzen unterschiedliche Gene und in unterschiedlichen Mengen. Diese Variation produziert viele verschiedene Arten von Gehirnzellen und trägt zur Komplexität neuronaler Schaltkreise bei. Zu lernen, wie sich diese Zelltypen auf molekularer Ebene unterscheiden, ist entscheidend für das Verständnis der Funktionsweise des Gehirns und die Entwicklung neuer Wege zur Behandlung neuropsychiatrischer Erkrankungen.
„Das menschliche Gehirn ist nicht homogen “, sagte der leitende Autor Bing Ren, PhD, Professor an der UC San Diego School of Medicine. „Es besteht aus einem äußerst komplexen Netzwerk von Neuronen und nicht-neuronalen Zellen, von denen jede unterschiedliche Funktionen erfüllt. Die Kartierung der verschiedenen Zelltypen im Gehirn und das Verständnis ihrer Zusammenarbeit werden uns letztendlich dabei helfen, neue Therapien zu entdecken, die gezielt wirken können.“ auf einzelne Zelltypen, die für bestimmte Krankheiten relevant sind.
In der neuen Studie analysierten Forscher mehr als 1,1 Millionen Gehirnzellen in 42 verschiedenen Gehirnregionen von drei menschlichen Gehirnen. Sie identifizierten 107 verschiedene Subtypen von Gehirnzellen und konnten Aspekte ihrer Molekularbiologie mit einer Vielzahl neuropsychiatrischer Erkrankungen in Verbindung bringen, darunter Schizophrenie, bipolare Störung, Alzheimer-Krankheit und schwere Depression . Forscher nutzen diese Daten dann, um Modelle für maschinelles Lernen zu erstellen, um vorherzusagen, wie bestimmte Sequenzvariationen in der DNA die Genregulation beeinflussen und zur Krankheit beitragen können.
Obwohl diese neuen Ergebnisse wichtige Informationen über das menschliche Gehirn und seine Pathologie liefern, sind Wissenschaftler mit der Kartierung des Gehirns noch lange nicht fertig. Im Jahr 2022 gründete die UC San Diego gemeinsam mit dem Salk Institute und anderen ein Multi-Omic Atlas Center of Human Brain Cells, dessen Ziel es ist, Zellen aus mehr als einem Dutzend menschlicher Gehirne zu untersuchen und Fragen darüber zu stellen, wie sich das Gehirn während der Entwicklung verändert. ein Leben lang und bei Krankheit.
„Die Ausweitung unserer Arbeit auf eine noch größere Detailebene in einer größeren Anzahl von Gehirnen wird uns dem Verständnis der Biologie neuropsychiatrischer Störungen und der Frage, wie sie rehabilitiert werden können, einen Schritt näher bringen“, sagte Ren.
Zu den Hauptautoren der Studie gehören: Yang Eric Li, Sebastian Preissl, Michael Miller, Zihan Wang, Henry Jiao, Chenxu Zhu, Zhaoning Wang, Yang Xie, Olivier Poirion, Colin Kern, Lin Lin, Qian Yang, Quan Zhu, Nathan Zemke. , Sarah Espinoza, Jingbo Shang und Allen Wang an der UC San Diego, Nicholas D. Johnson Antonio Pinto-Duarte, Wei Tian Nora Emerson, Julia Osteen, Jacinta Lucero, M. Margarita Behrens und Joseph R. Ecker am Salk Institute for Biological Studies , Kimberly Silett und Sten Linnarsson vom Karolinksa Institute Anna Marie Yanny, Julie Nyhus, Nick Dee, Tamara Casper, Nadiya Shapovalova, Daniel Hirschstein, Rebecca D. Hodge Trygve Bakken, Boaz Levi und Ed Lein vom Allen Institute for Brain Sciences und C . Dirk Keene von der University of Washington Seattle.
Die Studie wurde von den National Institutes of Health (Zuschüsse UM1MH130994, U01MH114812, U54HG012510 und S10 OD026929), der National Science Foundation (Zuschuss OIA-2040727) und der National Science Foundation (Zuschuss OIA-2040727) unterstützt. das Nancy and Buster Alford Endowment, die Life Sciences Research Foundation sowie Spenden von Google, Adobe und Teradata.















